丙肝治疗药物Ravidasvir的制备方法与流程
本发明涉及制药技术领域,具体地涉及丙肝治疗药物ravidasvir(asc16)的制备方法。
背景技术:
丙肝,全称为丙型病毒性肝炎,是一种由丙型肝炎病毒(hcv)感染引起的病毒性肝炎,主要经输血、针刺、吸毒等途径传播。据世界卫生组织的统计,全球丙肝病毒的感染率约为3%,估计约1.8亿人感染了丙肝病毒,每年新发丙型肝炎病例约3.5万例。丙型肝炎全球性流行,可导致肝脏慢性炎症坏死和纤维化,部分患者可发展为肝硬化甚至肝细胞癌(hcc)。丙肝对患者的健康和生命危害极大,已成为严重的社会和公共卫生问题。
世界著名的大型医药公司已经开发了多种治疗丙肝的药物。其中,presidio公司开发的ravidasvir(asc16,结构如以下式i所示)是一种高效的ns5a抑制剂,日前在中国已经进入临床阶段。
在presidio公司的专利申请wo2013/123092中,公开了asc16的制备方法。在该专利申请的制备方法的多个步骤中,公斤级的投料也需要采用柱层析进行纯化。柱层析纯化效率低,难以适应工业化大生产的要求。因此,开发新的ravidasvir的制备工艺,降低生产成本,提高生产效率,具有重要的社会意义和经济意义。
技术实现要素:
本发明中用到的缩写如下:
meoh:甲醇etoh:乙醇dcm:二氯甲烷
thf:四氢呋喃etoh.hcl:盐酸乙醇溶液
pd(dppf)cl2.dcm:pd(dppf)cl2二氯甲烷络合物
moc-l-缬氨酸:n-(甲氧羰基)-l-缬氨酸
hopo:2-羟基吡啶-n-氧化物
edci:1-(3-二甲氨基丙基)-3-乙基碳二亚胺
dipea:n,n-二异丙基乙胺
本发明的目的是提供一种改进的ravidasvir(式i所示化合物)的制备方法,该方法的工艺路线如下所示。
本发明提供的ravidasvir的制备方法包括以下步骤:
(1)式b4所示化合物与联硼酸频那醇酯在醋酸钾和pd(dppf)cl2二氯甲烷络合物(pd(dppf)cl2.dcm)存在下,在四氢呋喃中回流反应;反应结束后,经后处理得到式b5所示化合物;
(2)将四氢呋喃、式b5所示化合物、式c2所示化合物混合后,再加入碳酸钾水溶液、pd(dppf)cl2.dcm,回流反应;反应结束后,降至室温,分出有机相,减压浓缩;向浓缩产物加入乙酸乙酯和活性炭,搅拌,过滤,滤液减压浓缩后采用丙酮进行第一次丙酮结晶,过滤,得到式c3所示化合物的粗湿品;然后向式c3所示化合物的粗湿品加入丙酮进行第二次丙酮结晶,得到式c3所示化合物;
(3)将式c3所示化合物和乙醇混合,加入盐酸乙醇溶液反应,脱去叔丁氧羰基(boc保护基),得到式c4所示化合物;
(4)将n-(甲氧羰基)-l-缬氨酸(moc-l-缬氨酸)、2-羟基吡啶-n-氧化物(hopo)、1-(3-二甲氨基丙基)-3-乙基碳二亚胺(edci)和二氯甲烷混合,然后加入式c4所示化合物;滴加n,n-二异丙基乙胺(dipea)进行反应;反应结束后,加水萃取;将有机层减压浓缩,向浓缩产物加入乙酸乙酯和活性炭,加热搅拌,过滤;滤液减压浓缩至干;将浓缩产物用甲醇溶解,然后将所得甲醇溶液缓慢滴加入水中,析晶,得到式c5所示化合物;
(5)利用盐酸乙醇溶液使式c5所示化合物成盐。
在本发明方法的步骤(1)中,pd(dppf)cl2.dcm与联硼酸频那醇酯的质量比为1∶35。在本发明方法的步骤(1)中,所述后处理为:冷却,过滤;向滤液中加入活性炭,搅拌,过滤,滤液浓缩后,加入异丙醇搅拌溶清,然后减压蒸馏至干,再次加入异丙醇,升温至80~85℃,搅拌至少1小时;再缓慢降温至-5~0℃,且搅拌至少3小时;离心,并用异丙醇洗涤湿品,干燥,得到式b5所示化合物。优选地,洗涤滤饼所用的异丙醇的温度为-5~0℃。
在本发明方法的步骤(2)中,所述碳酸钾水溶液的浓度为0.5mol/l至1.0mol/l。
在本发明方法的步骤(2)中,pd(dppf)cl2.dcm与式b5所示化合物的质量比为1∶40。
在本发明方法的步骤(2)中,在第一次丙酮结晶得到式c3所示化合物的粗湿品后,先将式c3所示化合物的粗湿品用二氯甲烷溶清,然后减压蒸馏至干;然后向c3所示化合物的粗湿品加入丙酮进行第二次丙酮结晶,离心、干燥,得到式c3所示化合物。试验结果表明,增加这个操作步骤可以在后面的第二次丙酮结晶中除去更多的偶联杂质。
在本发明方法的步骤(3)和(5)中,所述盐酸乙醇浓度的浓度为12.5wt%。
在本发明方法的步骤(4)中,所述萃取还包括将有机层分别进行酸洗和碱洗,以及纯化水洗涤。优选地,所述酸洗和碱洗分别采用1wt%的柠檬酸水溶液与5wt%的碳酸氢钠水溶液,或1wt%的醋酸与5wt%的碳酸钠溶液进行。
优选地,在步骤(4)中,滤液减压浓缩至干,然后加入甲醇溶解、减压蒸馏至无液滴后再次加入甲醇,将浓缩产物用甲醇溶解。
在本发明提供的制备方法中,式c3所示化合物的制备过程中使用了丙酮进行两次结晶,经过两次丙酮结晶,可以除去成品中绝大部分的偶联杂质和其他微量杂质。在式c5所示化合物的制备过程中使用了甲醇进行重结晶。本发明提供的制备方法全程未采用柱层析操作。因此,本发明的工艺方法操作简单,生产周期短,溶剂成本低,更适合放大生产。
在发明的方法中,重结晶前先用活性炭除去重金属钯,取代现有技术中采用硅胶柱除去钯的方法,使得操作更加简单,其中步骤(1)和步骤(2)中pd(dppf)cl2.dcm的使用量降低到5wt%以下,大大节约了生产成本。同时,本发明的制备方法还避免了二氧六环的使用,对环境和操作人员更加友好。
在式c5所示化合物的制备反应中,本发明使用hopo作为缩合剂,研究表明其相对于hatu、hobt或tbtu等缩合剂,产生的异构体副产物更少,更有利于后续使用重结晶的方法进行纯化,收率明显提高,达到80-90%,即本发明的前后步骤相互协同,产生预料不到的技术效果。
与现有技术文献wo2013/123092相比,本发明的方法不仅更适合工业化大生产,而且有些步骤的收率与现有技术文献相当,有些步骤的收率还要比现有技术高出20%以上。在吨级的生产规模上,收率提高20%所产生的经济效益是巨大的。因此,本发明的方法是一种更加适于ravidasvir工业化大生产的生产方法。
附图说明
以下,结合附图来详细说明本发明的实施方案,其中:
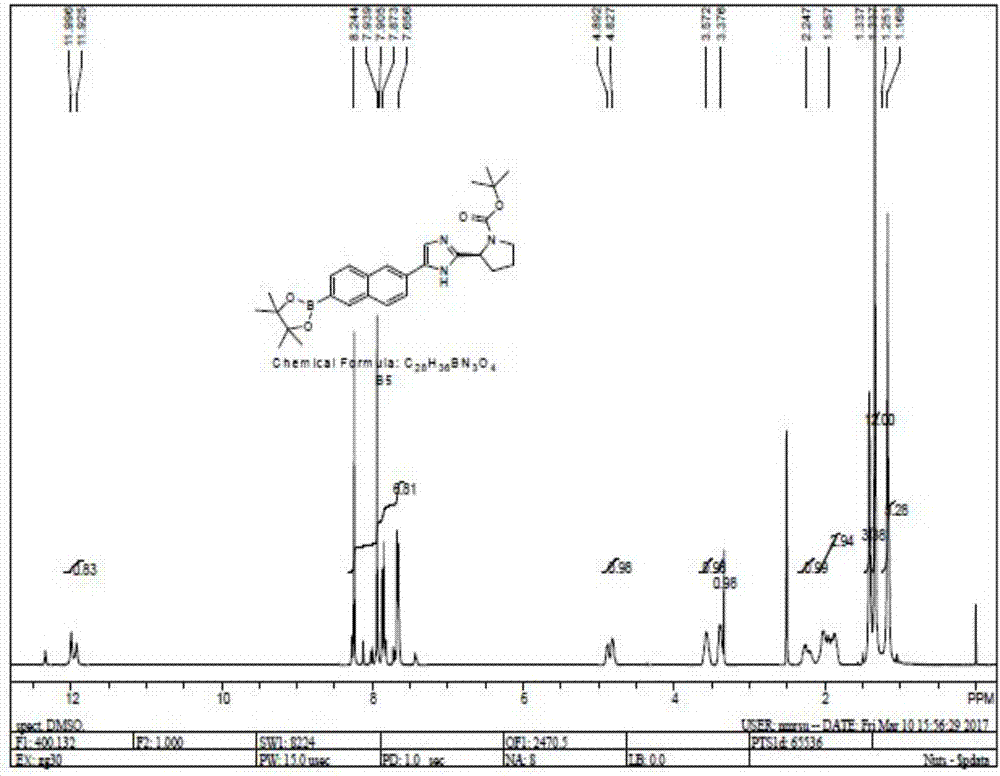
图1是式b5所示化合物的1hnmr图谱。
图2是式c3所示化合物的1hnmr图谱。
图3是式c4所示化合物的1hnmr图谱。
图4是式c5所示化合物的1hnmr图谱。
图5是化合物ravidasvir的1hnmr图谱。
具体实施方式
实施例1
式b5所示化合物的合成
在反应釜中加入1410kg四氢呋喃,150.0kgb4,100.5kg醋酸钾,94.5kg联硼酸频那醇酯,2.7kgpd(dppf)cl2.dcm;加热升温至回流(60~75℃)。搅拌至少12小时;hplc检测反应至反应完全(b4≤0.50%);冷却溶液至20~25℃,过滤并用四氢呋喃淋洗滤饼至淋洗液无色。滤液及淋洗液都合并至另一反应釜中,加入18.0kg活性炭,搅拌至少1h;过滤活性炭并用四氢呋喃淋洗滤饼至滤液无色;滤液减压蒸馏后加入477.0kg异丙醇,搅拌至少30分钟,减压蒸馏至干,以除去残留的thf,减少产物b5的损失。反应釜中再加入477.0kg异丙醇,升温至80~85℃,搅拌至少1小时;再缓慢降温至-5~0℃,且搅拌至少3小时;离心,并用353.0kg异丙醇(-5~0℃)洗涤得湿品;在45~55℃下真空干燥,得b5(收率70~80%)。
lcms(esi)m/z:490(m+1);1hnmr(400mhz,dmso-d6):δ12.0-11.9(m,1h),8.25-8.22(m,2h),7.95-7.90(m,2h),7.85-7.80(m,1h),7.72-7.65(m,2h),4.90-4.80(m,1h),3.65-3.50(m,1h),3.40-3.30(m,1h),2.30-2.15(m,1h),2.10-1.30(m,3h),1.45-1.35(m,3h),1.34(s,12h),1.17(m,6h)。
实施例2
式c3所示化合物的合成
在反应釜r-1中加入1200kg纯化水,85.0kg碳酸钾,搅拌溶清得碳酸钾水溶液(0.513mol/l);在反应釜r-2中加入1108kg四氢呋喃,120.0kgb5,90.0kgc2搅拌溶清;将碳酸钾水溶液从反应釜r-1转入至r-2中,加入3.0kgpd(dppf)cl2.dcm;升温至回流(65~75℃)搅拌至少12小时;取样分析至合格(c2≤0.10%);降温至20~25℃分层;减压蒸馏至无液滴;在反应釜r-2中加入2215kg乙酸乙酯,搅拌溶解,加入44.0kg活性炭,升温至65~70℃搅拌1~2小时;降温至20~25℃,过滤活性炭,用乙酸乙酯淋洗滤饼;减压蒸馏至无液滴后加入942kg丙酮,在20~30℃下搅拌6~12小时后离心,用共约185kg丙酮淋洗滤饼,得c3粗湿品;向反应釜r-2投入离心粗湿品和3323kg二氯甲烷,搅拌溶清,减压蒸馏至无液滴;反应釜中加入1938kg丙酮,在20~30℃下搅拌16~18小时后离心,用185kg丙酮漂洗滤饼,得c3精制湿品;在45~55℃下真空干燥,至干燥失重≤3.0%后降温卸料,得干品c3(收率45~60%)。
lcms(esi)m/z:649(m+1);1hnmr(400mhz,dmso-d6):δ12.4-12.3(m,1h),12.0-11.8(m,1h),8.30-7.52(m,10h),5.08-4.75(m,2h),3.68-3.58(m,2h),3.45-3.32(m,2h),2.37-1.82(m,8h),1.42-1.10(m,18h)。
实施例3
式c4所示化合物的合成
在反应釜中投入90.0kgc3,640kg乙醇;保持温度低于30℃(20~30℃),加入485kg(12.5%)盐酸乙醇溶液;在20~30℃下搅拌至少16小时,取样检测(c3≤0.10%)至合格;加入792kg四氢呋喃在20~30℃搅拌至少3h;离心得c4粗湿品;反应釜中投入离心粗湿品,401kg四氢呋喃和356kg乙醇,搅拌至少1h;离心得c4第一次打浆湿品;反应釜中投入第一次打浆湿品,793kg四氢呋喃,搅拌至少1h;离心得c4第二次打浆湿品;在50~55℃下真空干燥,至干燥失重≤5.0%后降温卸料,得c4干品(收率70~90%)。
lcms(esi)m/z:449(m+1);1hnmr(400mhz,dmso-d6):δ10.8-10.6(m,br,1h),10.3-9.90(m,br,1h),8.66(s,1h),8.40-8.30(m,2h),8.20-7.95(m,5h),7.92-7.85(m,2h),5.25-5.12(m,2h),3.48-3.60(m,4h),2.62-2.51(m,3h),2.50-2.45(m,1h),2.31-2.20(m,2h),2.17-2.00(m,2h).
实施例4
式c5所示化合物的合成
向反应釜中投入42.6kgmoc-l-缬氨酸,27.0kghopo,46.2kgedci,1600kg二氯甲烷,在20~25℃下搅拌溶清,向反应釜中加入60.0kgc4;在30℃以下缓慢滴加92kgdipea;在25~30℃下搅拌至少2h;取样分析(c4≤0.10%)至合格;向反应釜r-1中加入1200kg纯化水搅拌分层;有机层分别用1%的柠檬酸溶液(1200kg),5%的碳酸氢钠(600kg×2),纯化水(600kg×2)分别洗涤,减压蒸馏至无液滴;反应釜中加入540kg乙酸乙酯及6.0kg活性炭,加热升温至60~65℃搅拌至少2h;降温至25~30℃过滤并用270kg乙酸乙酯淋洗;减压蒸馏至无液滴;向反应釜r-1中加入200kg甲醇,搅拌至少30min;减压蒸馏至无液滴后再加入163kg甲醇,搅拌至溶清得c5/甲醇溶液,反应釜中加入1200kg纯化水并将c5/甲醇溶液缓慢滴加入反应釜中,搅拌至少2h;离心,用纯化水漂洗得c5离心湿品;在45~55℃下真空干燥得c5干品(收率80~90%)。
lcms(esi)m/z:763(m+1);1hnmr(400mhz,dmso-d6):δ12.6-12.3(m,2h),7.74(s,2h),7.65-7.60(m,1h),7.55-7.48(m,2h),7.42-7.38(m,2h),7.30-7.22(m,3h),5.00-4.82(m,2h),3.58-3.52(m,2h),3.42-3.33(m,2h),2.30-2.18(m,2h),2.02-1.30(m,6h),1.39(s,6h),1.06(m,12h)。
实施例5
成盐酸盐
向反应釜中加入60.0kgc5,142kg乙醇,211kg乙酸正丁酯;搅拌至溶清,加入43.6kg(12.5%)的盐酸乙醇溶液;搅拌至少30min,将料液过滤至结晶釜中,并用57kg乙醇和53kg乙酸正丁酯淋洗过滤系统;缓慢升温至60~65℃加入0.3kgasc16晶种,在60~65℃下保温搅拌至少24h;离心,用190kg乙酸正丁酯漂洗得到的asc16离心湿品;在55~60℃下真空干燥得asc16干品,收率80%。
lcms(esi)m/z:763(m+1);1hnmr(400mhz,dmso-d6):δ15.5(s,br,2h),15.0(s,1h),8.62(s,1h),8.35(s,1h),8.26(s,1h),8.20-8.17(m,1h),8.14-8.12(m,1h),8.09-8.05(m,2h),8.01-7.97(m,2h),7.94-7.90(m,1h),7.37-7.30(m,2h),5.36-5.32(m,1h),5.28-5.24(m,1h),4.21-4.13(m,2h),4.11-4.04(m,2h),3.92-3.82(m,2h),3.56(s,6h),2.50-2.35(m,2h),2.35-2.30(m,1h),2.30-1.90(m,7h),1.00-0.75(m,12h)。
- 还没有人留言评论。精彩留言会获得点赞!