一种分步催化制备高纯度棓丙酯的方法与流程
本发明涉及有机合成技术领域,尤其涉及一种分步催化制备高纯度棓丙酯的方法。
背景技术:
棓丙酯化学名称为没食子酸丙酯,分子式c10h12o5,分子量212.20,具有抑制(血栓素a2)txa2引起的血小板聚集作用;可降低全血粘度和血浆比粘度,加快红细胞电泳速度,也可松弛血管平滑肌,增加冠状动脉的血流量;对心肌缺血有明显的保护作用。在扩张脑血管,改善脑循环的同时,还可通过提高脑组织sod,gsh-px和cat活性,减轻脑组织的脂质过氧化反应,抑制脑水肿形成,并保护脑组织na+,k+-atp酶活性,有效地减轻脑缺血再灌注损伤,对防止脑缺血再灌注损伤有良好的应用前景。
棓丙酯的制备一般以没食子酸为原料,进行酯化反应。由于没食子酸和产物棓丙酯化学性质相近,因此少量反应不完全的没食子酸在反应结束后较难除去,给纯化带来较大的难度,使得棓丙酯的纯度受到影响。
随着人们生活水平不断提升,用药安全受到越来越多的重视,对药品药典杂质标准要求也相应提高,2010版药典规定棓丙酯检测项有关物质中主要原料没食子酸杂质不得大于0.1%,总杂不得大于0.5%,这对很多还是按照普通单步催化酯化工艺生产的棓丙酯厂家产生了很大的挑战,很难按照普通的合成工艺生产出没食子酸含量合格的棓丙酯。
技术实现要素:
有鉴于此,本发明要解决的技术问题在于提供一种分步催化制备高纯度棓丙酯的方法,制备的棓丙酯具有较高的纯度。
为解决以上问题,本发明提供了一种分步催化制备高纯度棓丙酯的方法,包括以下步骤:
以没食子酸和正丙醇为原料,依次在高氯酸和氨基磺酸的催化作用下,或者在对甲苯磺酸和氨基磺酸的催化作用下,进行反应,得到棓丙酯。
本发明采用高氯酸和氨基磺酸,或者对甲苯磺酸和氨基磺酸,进行分步催化,使没食子酸和正丙醇的酯化反应充分,没有原料没食子酸残留。
上述反应的方程式如下:
在本发明的一些具体实施例中,采用一水没食子酸为原料。
具体的,以上分步催化的方法包括以下步骤:
以没食子酸和正丙醇为原料,在高氯酸或者对甲苯磺酸的催化作用下,回流条件下,进行第一次催化反应;
待反应体系出水量达到理论值,加入氨基磺酸,进行第二次催化反应,得到棓丙酯。
所述没食子酸和正丙醇中,优选正丙醇过量,优选的,所述没食子酸和正丙醇的摩尔比为1:(2~3)。
所述高氯酸或者对甲苯磺酸和没食子酸的摩尔比优选为(0.1~0.3):1,进一步优选为0.2:1。
所述反应的带水剂优选为苯。
本发明对所述苯的用量并无特殊限定,可以根据本领域技术人员的一般经验进行调节。
所述第一次催化反应的温度优选为80~90℃,反应时间优选为12~14h。
第一次催化反应过程中,反应体系由浆状转为暗红色溶液。
本发明优选的,在进行回流反应的同时,采用分水器收集并测量苯带出的水。当反应体系出水量达到理论值时,加入氨基磺酸,进行第二次催化反应,得到棓丙酯。
所述氨基磺酸和没食子酸的摩尔比优选为(0.05~0.1):1,进一步优选为0.05:1。
本发明优选的,加入氨基磺酸时降低体系温度,优选的,加入氨基磺酸时,体系的温度为50~60℃。氨基磺酸加入完毕,体系温度恢复至回流温度。
优选的,所述第二次催化反应的温度优选为80~90℃,反应时间优选为1~2h。
反应结束后,反应体系浓缩至总体积的一半,得到棓丙酯粗品浓缩液。
本发明优选的,对所述棓丙酯粗品进行提纯,优选具体的:
加入水进行结晶,固体过滤、水洗涤至中性,活性炭脱色,水重结晶,得到棓丙酯纯品。
提纯步骤采用的水优选为纯化水。
经检测,本发明提供的上述方法合成出的棓丙酯符合2015版药典质量要求,且纯度高达99.9%以上,未检出没食子酸杂质峰。
附图说明
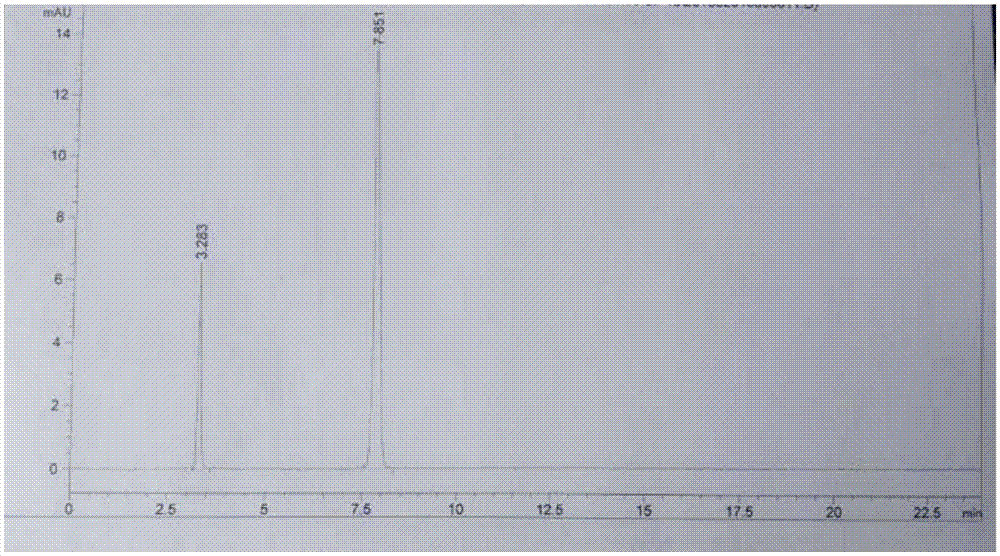
图1为没食子酸和棓丙酯混合溶液的hplc图谱;
图2为本申请实施例2制备的棓丙酯的hplc图谱。
具体实施方式
为了进一步说明本发明,下面结合实施例对本发明提供的分步催化制备高纯度棓丙酯的方法进行详细描述。
实施例1
准确称取一水没食子酸37.6g(0.2mol),加入500ml三口烧瓶中,依次量取高氯酸1.6ml(0.02mol),苯40ml,取正丙醇45ml(0.6mol),加入三口烧瓶中,三口烧瓶置于1l电加热套中,三口烧瓶一端口插入温度计,另一端口接分水器,分水器上端接回流管,中间口用长柄夹固定,接带密封四氟搅拌桨,开启搅拌电机、电加热套及冷凝水,保持温度80℃~90℃搅拌回流反应,反应12小时~14小时左右。
当分水器中收集了约7.2ml(0.4mol)冷凝水,加入0.97g(0.01mol)氨基磺酸,继续搅拌回流反应1小时后减压浓缩至总体积的一半,得棓丙酯粗品浓缩液。
棓丙酯浓缩液经纯化水结晶后,在粗品晶体中未测出没食子酸原料,棓丙酯粗品用纯化水洗涤至中性,活性炭脱色,纯化水重结晶后过滤烘干,得精品棓丙酯28.4g,收率约66.98%,纯度99.6%。
实施例2
准确称取一水没食子酸(0.2mol)37.6g,加入500ml三口烧瓶中,依次量取高氯酸3.2(0.04mol)ml,苯40ml,取正丙醇45ml(0.6mol),加入三口烧瓶中,三口烧瓶置于1l电加热套中,三口烧瓶一端口插入温度计,另一端口接分水器,分水器上端接回流管,中间口长柄夹夹住,接四氟搅拌桨,开启搅拌电机,电加热套及冷凝水,保持温度80℃~90℃搅拌回流反应,反应12小时~14小时左右。
当分水器中收集了约7.2ml(0.4mol)冷凝水,加入0.97g(0.01mol)氨基磺酸,继续搅拌回流反应1小时后减压浓缩至总体积的一半,得棓丙酯粗品浓缩液。
棓丙酯浓缩液经纯化水结晶后,在粗品晶体中未测出没食子酸原料,棓丙酯粗品用纯化水洗涤至中性,活性炭脱色,纯化水重结晶后过滤烘干,得精品棓丙酯34.6g,收率约81.60%,纯度99.9%。
实施例3
准确称取一水没食子酸(0.2mol)37.6g,加入500ml三口烧瓶中,依次量取高氯酸4.8(0.06mol)ml,苯40ml,取正丙醇45ml(0.6mol),加入三口烧瓶中,三口烧瓶置于1l电加热套中,三口烧瓶一端口插入温度计,另一端口接分水器,分水器上端接回流管,中间口长柄夹夹住,接四氟搅拌桨,开启搅拌电机,电加热套及冷凝水,保持温度80℃~90℃搅拌回流反应,反应12小时~14小时左右。
当分水器中收集了约7.2ml(0.4mol)冷凝水,加入0.97g(0.01mol)氨基磺酸,继续搅拌回流反应1小时后减压浓缩至总体积的一半,得棓丙酯粗品浓缩液。
棓丙酯浓缩液经纯化水结晶后,在粗品晶体中未测出没食子酸原料,棓丙酯粗品用纯化水洗涤至中性,活性炭脱色,纯化水重结晶后过滤烘干,得精品棓丙酯31.3g,收率约73.80%,纯度99.6%。
实施例4
准确称取一水没食子酸(0.2mol)37.6g,加入500ml三口烧瓶中,依次量取高氯酸3.2(0.04mol)ml,苯40ml,取正丙醇45ml(0.6mol),加入三口烧瓶中,三口烧瓶置于1l电加热套中,三口烧瓶一端口插入温度计,另一端口接分水器,分水器上端接回流管,中间口长柄夹夹住,接四氟搅拌桨,开启搅拌电机,电加热套及冷凝水,保持温度80℃~90℃搅拌回流反应,反应12小时~14小时左右。
当分水器中收集了约7.2ml(0.4mol)冷凝水,加入1.94g(0.02mol)氨基磺酸,继续搅拌回流反应1小时后减压浓缩至总体积的一半,得棓丙酯粗品浓缩液。
棓丙酯浓缩液经纯化水结晶后,在粗品晶体中未测出没食子酸原料,棓丙酯粗品用纯化水洗涤至中性,活性炭脱色,纯化水重结晶后过滤烘干,得精品棓丙酯32.1g,收率约75.70%,纯度99.7%。
比较例1
准确称取一水没食子酸37.6g(0.2mol),加入500ml三口烧瓶中,依次量取高氯酸3.2ml(0.04mol),苯40ml,取正丙醇45ml(0.6mol),加入三口烧瓶中,三口烧瓶置于1l电加热套中,三口烧瓶一端口插入温度计,另一端口接分水器,分水器上端接回流管,中间口长柄夹夹住,接四氟搅拌桨,开启搅拌电机,电加热套及冷凝水,保持温度80℃~90℃搅拌回流反应,反应12小时~14小时左右。
当分水器中收集了约7.2ml(0.4mol)冷凝水,减压浓缩至总体积的一半,得棓丙酯粗品浓缩液。
棓丙酯浓缩液经纯化水结晶后,在粗品晶体中测出没食子酸原料约0.25%色谱吸收,棓丙酯粗品用纯化水洗涤至中性,活性炭脱色,纯化水重结晶后过滤烘干,得精品棓丙酯27.1g,收率约63.920%,纯度99.1%。
实施例5
准确称取一水没食子酸37.6g(0.2mol),3.8g(0.02mol)一水对甲苯磺酸,加入500ml三口烧瓶中,依次量取苯40ml,取正丙醇45ml(0.6mol),加入三口烧瓶中,三口烧瓶置于1l电加热套中,三口烧瓶一端口插入温度计,另一端口接分水器,分水器上端接回流管,中间口长柄夹夹住,接四氟搅拌桨,开启搅拌电机,电加热套及冷凝水,保持温度80℃~90℃搅拌回流反应,反应12小时~14小时左右。
当分水器中收集了约7.6ml(0.42mol)冷凝水,加入0.97g氨基磺酸,继续搅拌回流反应1小时后减压浓缩至总体积的一半,得棓丙酯粗品浓缩液。
棓丙酯浓缩液经纯化水结晶后,在粗品晶体中未测出没食子酸原料约的色谱吸收,棓丙酯粗品用纯化水洗涤至中性,活性炭脱色,纯化水重结晶后过滤烘干,得精品棓丙酯28.9g,收率约68.16%,纯度99.6%。
实施例6
准确称取一水没食子酸37.6g(0.2mol),7.6g(0.04mol)一水对甲苯磺酸,加入500ml三口烧瓶中,依次量取苯40ml,取正丙醇45ml(0.6mol),加入三口烧瓶中,三口烧瓶置于1l电加热套中,三口烧瓶一端口插入温度计,另一端口接分水器,分水器上端接回流管,中间口长柄夹夹住,接四氟搅拌桨,开启搅拌电机,电加热套及冷凝水,保持温度80℃~90℃搅拌回流反应,反应12小时~14小时左右。
当分水器中收集了约7.9ml(0.44mol)冷凝水,加入0.97g氨基磺酸,继续搅拌回流反应1小时后减压浓缩至总体积的一半,得棓丙酯粗品浓缩液。
棓丙酯浓缩液经纯化水结晶后,在粗品晶体中未测出没食子酸原料的色谱吸收,棓丙酯粗品用纯化水洗涤至中性,活性炭脱色,纯化水重结晶后过滤烘干,得精品棓丙酯32.2g,收率约75.95%,纯度99.7%。
实施例7
准确称取一水没食子酸37.6g(0.2mol),11.4g(0.06mol)一水对甲苯磺酸,加入500ml三口烧瓶中,依次量取苯40ml,取正丙醇45ml(0.6mol),加入三口烧瓶中,三口烧瓶置于1l电加热套中,三口烧瓶一端口插入温度计,另一端口接分水器,分水器上端接回流管,中间口长柄夹夹住,接四氟搅拌桨,开启搅拌电机,电加热套及冷凝水,保持温度80℃~90℃搅拌回流反应,反应12小时~14小时左右。
当分水器中收集了约8.3ml(0.46mol)冷凝水,加入0.97g氨基磺酸,继续搅拌回流反应1小时后减压浓缩至总体积的一半,得棓丙酯粗品浓缩液。
棓丙酯浓缩液经纯化水结晶后,在粗品晶体中未测出没食子酸原料的色谱吸收,棓丙酯粗品用纯化水洗涤至中性,活性炭脱色,纯化水重结晶后过滤烘干,得精品棓丙酯29.3g,收率约69.10%,纯度99.5%。
实施例8
准确称取一水没食子酸37.6g(0.2mol),7.6g(0.04mol)一水对甲苯磺酸,加入500ml三口烧瓶中,依次量取苯40ml,取正丙醇45ml(0.6mol),加入三口烧瓶中,三口烧瓶置于1l电加热套中,三口烧瓶一端口插入温度计,另一端口接分水器,分水器上端接回流管,中间口长柄夹夹住,接四氟搅拌桨,开启搅拌电机,电加热套及冷凝水,保持温度80℃~90℃搅拌回流反应,反应12小时~14小时左右。
当分水器中收集了约7.9ml(0.44mol)冷凝水,加入1.94g(0.02mol)氨基磺酸,继续搅拌回流反应1小时后减压浓缩至总体积的一半,得棓丙酯粗品浓缩液。
棓丙酯浓缩液经纯化水结晶后,在粗品晶体中未测出没食子酸原料的色谱吸收,棓丙酯粗品用纯化水洗涤至中性,活性炭脱色,纯化水重结晶后过滤烘干,得精品棓丙酯30.0g,收率约70.75%,纯度99.6%。
比较例2
准确称取一水没食子酸37.6g(0.2mol),7.6g(0.04mol)一水对甲苯磺酸,加入500ml三口烧瓶中,依次量取苯40ml,取正丙醇45ml(0.6mol),加入三口烧瓶中,三口烧瓶置于1l电加热套中,三口烧瓶一端口插入温度计,另一端口接分水器,分水器上端接回流管,中间口长柄夹夹住,接四氟搅拌桨,开启搅拌电机,电加热套及冷凝水,保持温度80℃~90℃搅拌回流反应,反应12小时~14小时左右。
当分水器中收集了约7.9ml(0.44mol)冷凝水,减压浓缩至总体积的一半,得棓丙酯粗品浓缩液。
棓丙酯浓缩液经纯化水结晶后,在粗品晶体中测出没食子酸原料约0.30%的色谱吸收,棓丙酯粗品用纯化水洗涤至中性,活性炭脱色,纯化水重结晶后过滤烘干,得精品棓丙酯26.9g,收率约63.44%,纯度99.1%。
由上述实施例及比较例可知,本发明采用高氯酸和氨基磺酸,或者对甲苯磺酸和氨基磺酸体系,进行分步催化反应,制备的棓丙酯具有较高的纯度。
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!