一种CHO细胞基因组内NW_006880285-1稳定表达蛋白质的应用的制作方法
本发明涉及基因技术领域,尤其是涉及一种用于稳定表达蛋白质的cho细胞重组基因。
背景技术:
中国仓鼠卵巢细胞(chinesehamsterovarycell,cho)作为生物制药领域的主力细胞系,已经被开发出许多不同种类的cho细胞系,包括可以用来扩大基因拷贝数的细胞系。然而,转基因拷贝数的增多与目的蛋白质产率提高并不是明确的正相关关系。且即使蛋白质表达增多,多数cho细胞的表达水平不稳定。当前普遍使用的构建稳定转染细胞的方法耗时耗力,主要是因为需要重复大量的单克隆筛选过程,所以当前在细胞系构建领域普遍期待可以开发出一种能在短时间内,获得高表达及稳定表达的细胞的方法,并且能够确保构建出来的重组细胞系与传统方法相比,有相同的质量水平,以确保监管机构的批准。
传统构建外源蛋白质表达细胞系的方法是通过外源基因随机整合到细胞基因组上,再经过系列的高表达单克隆细胞筛选,获得外源蛋白质高表达细胞系。由于位点效应差异的多样性,随机整合产生的重组细胞表达水平各异,因此需要后期花费很长的时间和很多的步骤,用于挑选高表达单克隆细胞。通过随机整合获得的单克隆细胞无法保证多肽/蛋白质在细胞传代中稳定表达,每进行一次重组细胞构建均需要反复进行单克隆筛选,增加了生物制药的研发成本。
位点效应妨碍着传统随机整合构建重组细胞系的效率,反复的高表达单克隆筛选费时费力,且成本昂贵。如何克服位点效应,利用定点整合技术,快速高效地获得稳定表达单克隆细胞已经在学术界被讨论了多年,一直未有突破性进展。
技术实现要素:
针对现有技术存在的上述问题,本发明申请人提供了一种cho细胞基因组内某位点稳定表达蛋白质的应用。本发明申请在cho细胞基因组内的固定位置,导入不同蛋白质基因,并进行稳定表达;此外在实现该定点整合的过程中,无需多次挑选单克隆以获得更高的表达细胞株,节省了大量的时间。
本发明的技术方案如下:
一种cho细胞基因组内某位点稳定表达蛋白质的应用,所述cho细胞基因组内用于稳定表达蛋白质的位点位于cho细胞基因nw_006880285.1的第1235357碱基处;
所述位点附近1235284-1235429范围内可被crispr/cas9技术识别的5'nnnnnnnnnnnnnnnnnnnnngg3'为靶序列。
所述蛋白质为分子量小于160kda的蛋白质。
所述蛋白质为多肽、功能性蛋白质、抗体、融合蛋白质中的一种。
所述靶序列为cho细胞基因nw_006880285.1的第1235357碱基处上游第1235285-1235307碱基处;所述靶序列为5’-gaaagaaggtctgatatcaaagg-3’。
优选方案,所述靶序列为5’-aaagaaggtctgatatcaaaggg-3’。
优选方案,所述靶序列为5’-cctcactagtacacgcaccatgg-3’。
优选方案,所述靶序列为5’-tagcttgctcacagtagcacagg-3’。
优选方案,所述靶序列为5’-cttgctcacagtagcacaggagg-3’。
优选方案,所述靶序列为5’-gctcacagtagcacaggaggagg-3’。
优选方案,所述靶序列为5’-ctcacagtagcacaggaggaggg-3’。
优选方案,所述靶序列为5’-gcaagctacatagttacccatgg-3’。
优选方案,所述靶序列为5’-ccatggtgcgtgtactagtgagg-3’。
优选方案,所述靶序列为5’-caacttttagctacattccttgg-3’。
优选方案,所述靶序列为5’-aggaatgtagctaaaagttgagg-3’。
一种含有所述用于表达蛋白质的重组供体载体。
所述的重组供体载体为cho细胞表达的载体。
所述的重组供体载体的制作方法为:将蛋白基因插入到质粒5’arm和3’arm中间的区域,使该核苷酸序列位于启动子的下游并受其调控,得到重组cho细胞表达质粒。
所述启动子为:cmv(人类巨细胞病毒来源的强哺乳动物表达启动子)、ef-1a(人延长因子1α来源的强哺乳动物表达启动子)、sv40(猿猴空泡病毒40来源的哺乳动物表达启动子)、pgk1(磷酸甘油酸酯激酶基因来源的哺乳动物启动子)、ubc(人类泛素c基因来源的哺乳动物启动子)、humanbetaactin(β-肌动蛋白基因来源的哺乳动物启动子)、cag(强杂交哺乳动物启动子)中的一种。
一种用于稳定表达蛋白质的cho重组细胞系。
一种cho细胞基因表达蛋白质的方法,其特征在于,包括以下步骤:
(1)用所述的重组供体载体转化cho细胞,得重组cho细胞;
(2)重组cho细胞在平板上培养,收集上清检测表达水平,以及对贴壁重组cho细胞进行悬浮驯化;
(3)悬浮驯化的重组cho细胞在小摇瓶内进行培养,并检测蛋白质的表达水平。
本发明申请还提供一种cho细胞基因组中稳定表达位点的选择:
1)构建带荧光标签的慢病毒,并推算其滴度。将igk-luc基因整合到plvx-cmv-mcs-t2a-zsgreen载体的多克隆位点后,再同时利用pspax2、pmd2g两个质粒进行三质粒转染至hek-293t细胞,于48小时及72小时两次取细胞上清液,超速离心上清液,获取慢病毒。
2)将cho细胞铺在6孔板上培养过夜,第二天将慢病毒稀释并以较低的moi(moi<1)(每个细胞对应病毒颗粒)来感染cho细胞,感染96小时后,通过流式细胞分选仪分选,将荧光强度最亮的部分细胞直接接种到96孔板内。连续培养一周后,细胞长成单克隆集落。以荧光显微镜观察cho细胞,标注形态正常且最亮的集落,待细胞数量到达要求后转移到24孔板内扩大培养。培养细胞达汇合度近90%时再转移到6孔板内培养,最后再扩大至10cm的培养皿中培养,冻存一部分细胞,剩余细胞继续扩大培养。
3)利用染色体步移技术lenti-xintegrationsiteanalysiskit(clontech:631263)寻找到慢病毒所有的cho细胞基因整合位点。
以荧光强度最亮、细胞形态和生长速度正常的若干个细胞系为材料,采用adrai,sspi,hpai三个限制性内切酶对基因组dna进行过夜酶切。其中基因组dna有2.5μg,限制性内切酶有80u,配成100μl反应体系。置于37℃下过夜酶切(16-18小时)。
利用dna回收试剂盒将酶切产物纯化回收。将酶切后的基因组dna4.8μl,加上1.9μl染色体步移接头genomewalkeradaptor(25μm)和0.5μlt4连接酶,配成8μl体系进行连接实验。置于16℃环境下,进行过夜连接。将连接体系70℃下加热5分钟,使连接酶失活。各个体系内加入32μltebuffer配置成相应的文库。
对文库进行2轮巢式pcr,将ltr区域同临近的基因组区域扩增出来。相关的pcr反应操作步骤可以参照lenti-xintegrationsiteanalysiskit(clontech:631263)试剂盒说明书进行。
最后将pcr产物进行电泳,将主要条带切胶回收后送测序。在获得每个细胞系所有慢病毒整合信息后,选择只有单拷贝慢病毒整合的cho细胞系的相关信息,将其序列信息在blast上同cho-k1基因组信息加以比对,找出高表达的整合位点。
本发明有益的技术效果在于:
本发明采用定点整合方法,通过把目的基因定点整合到稳定表达区域,能很好的克服随机整合带来的整合位点不确定性的问题,有效的规避了反复多轮的高表达单克隆筛选,能有效的减少生物制药构建稳定表达细胞系的研发时间,降低成本。
本发明在cho细胞基因的定点位置,导入蛋白质基因,并进行稳定表达。
附图说明
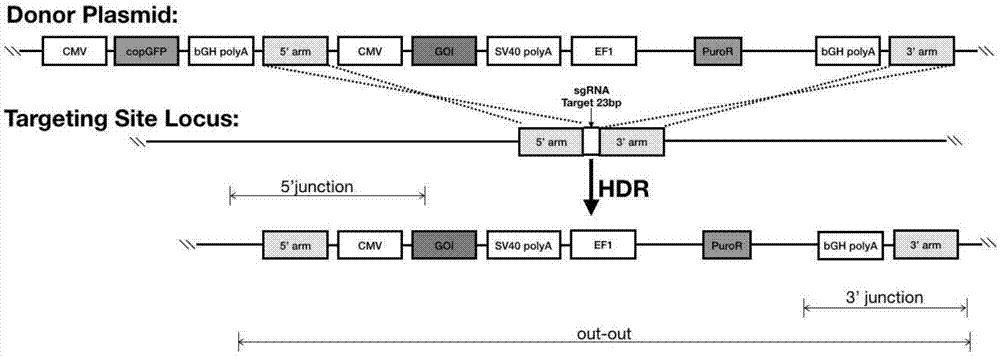
图1为本发明示意图;
图2为导入nggh75kda基因的cho细胞基因鉴定结果;
图3为oopcr_fwd及oopcr_rev对导入nggh75kda基因的cho细胞基因进行的测序;
图4为不同传代下的细胞表达hsa的情况;
图5为不同传代下的细胞表达nggh的情况;
图6为每日每个重组cho细胞分泌抗体蛋白的质量情况。
具体实施方式
下面结合附图和实施例,对本发明进行具体描述。
图1为用于整合到该位点的供体质粒示意图和如何通过同源重组定点整合到该位点的模拟示意图。其中goi为我们的目的基因,其通过5’arm和3’arm两个同源重组臂,在4μg/ml的puromycin筛选压力下,定点整合到靶点位置。此外5’arm上游的序列为负筛选标签,可以用来去除随机整合的单克隆细胞,确保最终获得的为定点整合的重组cho细胞系。
实施例1
高表达位点的选择;
nw_006880285.1,1235357碱基处整合,zsgreen1基因被整合在了该处。对于这个荧光细胞,进行了不少于50代的传代培养,通过流式细胞仪检测其荧光的表达水平。第50代荧光细胞仍有较好的绿色荧光蛋白表达水平。荧光信号在细胞的传代过程中被稳定的保留了。
此外对于这个荧光细胞还进行了悬浮驯化,并对其悬浮驯化后的荧光蛋白表达水平利用流式细胞仪再次进行了检测。检测结果显示,悬浮达到50代的重组cho细胞中,超过95%以上的细胞在悬浮后依然保持了绿色荧光蛋白的表达,因此可以认为该位点是极其稳定,不会因细胞传代丢失定点整合的荧光蛋白基因。
实施例2
具体靶点的选择;
根据就近原则,将序列:
5’cctcctcctgtgctactgtgagcaagctacatagttacccatggtgcgtgtactagtgaggtgattgattgacagactagtagaagcaca-cacctcaacttttagctacattccttggtctccctttgatatca-gaccttctttc3’
输入crisrprater系统中,预测并挑选出脱靶效率较低的靶序列。其中参数设置如下所示:1)ngg后开头15bp的最大错配碱基数为0;2)ngg后全部21bp的错配碱基数为2。
根据如上操作后,根据其评分选择分数为0.72的如下序列作为靶序列:
5’-gaaagaaggtctgatatcaaagg-3’;
根据crisprater系统,lowefficacy(score<0.56);mediumefficacy(0.56<=score<=0.74);highefficacy(score>0.74)。
根据crisprater评定系统,位点nw_006880285.1附近1235284-1235429范围内所有靶序列均获得了0.56以上的分数,均处于中等有效或高效范围,可作为crispr/cas9技术识别的5'nnnnnnnnnnnnnnnnnnnnngg3'靶序列。
实施例3启动子的选择
将不同的启动子替换上述cmv(人类巨细胞病毒来源的强哺乳动物表达启动子)启动子位置处,包含了ef-1a(人延长因子1α来源的强哺乳动物表达启动子)、sv40(猿猴空泡病毒40来源的哺乳动物表达启动子)、pgk1(磷酸甘油酸酯激酶基因来源的哺乳动物启动子)、ubc(人类泛素c基因来源的哺乳动物启动子)、humanbetaactin(β-肌动蛋白基因来源的哺乳动物启动子)或cag(强杂交哺乳动物启动子)等常见启动子。经检测,以上启动子均能调控下游的人血清白蛋白(hsa)基因序列,并表达出相应的hsa蛋白。
实施例4
将人血清白蛋白基因(hsa,68kda)定点整合在特定位点:为了后期进行构建crispr/cas9介导的同源重组,需要构建sgrna和donorplasmid,方法如下:
1、sgrna的构建,首先合成如下序列
sgrna-1fwd5’tttggaaagaaggtctgatatcaagt3’
sgrna-1rev5’taaaacttgatatcagaccttctttc3’
1)将psk-u6-grna质粒进行bbsi酶切,将切后的载体回收;
2)合成的片段退火成带有粘性末端的双链
水浴95℃5min,水浴锅中自然降至室温;
4)连接,转化;
5)挑取克隆,pcr鉴定,使用的鉴定引物为:
m13-合成的r的引物,可以扩增出条带的为阳性克隆。
2、donorplasmid构建
donorplsmid具体信息如图1所示:除goi之外,其余部分均为合成;靶点上下游600bp序列为donorplasmid的左右同源臂序列信息,goi为hsa基因通过已有的诺维赞公司c115试剂盒完成hsa整合到donorplasmid上这个过程。
3、将cas9(丹麦科技大学dr.helenefkildegaard捐赠)、sgrna及donorplasmid3个质粒共同转染37℃、5%co2条件下培养的cho细胞,三种质粒摩尔比为1:1:1,转染试剂为lipofectamine3000(thermofisherscientif-ic),具体转染方法参考说明书,之后细胞添加4μg/mlpuromycin进行筛选,过程共计10天;再用mofloxdpfacsmachine(beckmancoulter)进行单克隆细胞分选,选择不含任何荧光的细胞,接种到96孔板内。
4、细胞生长2周后,取一部分进行鉴定,通过做pcr鉴定,做5’junctionpcr,3’junctionpcr及out-outpcr进行鉴定。将阳性细胞保留。
实施例5
将胰高血糖素样肽-1-人血清白蛋白融合蛋白基因(nggh,75kda)定点整合在特定位点:为了后期进行构建crispr/cas9介导的同源重组,需要构建sgrna和donorplasmid,方法如下:
1、构建sgrna同实施例4。
2、donorplasmid构建。
donorplsmid具体信息如图1所示:除goi之外,其余部分均为合成;靶点上下游600bp序列为donorplasmid的左右同源臂序列信息,goi为nggh基因通过已有的诺维赞公司c115试剂盒完成nggh整合到donorplasmid上这个过程。
3、将cas9、sgrna及donorplasmid3个质粒共同转染37℃、5%、co2条件下培养的cho细胞,三种质粒摩尔比为1:1:1,转染试剂为lipofectamine3000(thermofisherscientific),具体转染方法参考说明书。之后细胞添加4μg/mlpuromycin进行筛选,过程共计10天。再用mofloxdpfacsmachine(beckmancoulter)进行单克隆细胞分选,选择不含任何荧光的细胞,接种到96孔板内。
4、细胞生长2周后,取一部分进行鉴定。通过做pcr鉴定,做5’junctionpcr,3’junctionpcr及out-outpcr进行鉴定。将阳性细胞保留。
图2为导入nggh75kda基因的cho基因鉴定结果,其中泳道1-3为三个单克隆细胞的5’jucntionpcr结果,泳道4-6为3’junctionpcr结果,均有显著条带,证实有基因敲入。
图3为利用oopcr_fwd及oopcr_rev进行测序,确定交界处(5’junction及3’junction的5’上游处与基因组的交界处)的序列准确,测序结果验证了goi被精准的插入到了靶点位置区域。
实施例6
将抗体基因(avastin,160kda)定点整合在特定位点:为了后期进行构建crispr/cas9介导的同源重组,需要构建sgrna和donorplasmid,方法如下:
1、sgrna构建同实施例4。
2、donorplasmid构建。
donorplsmid具体信息如图1所示:除goi之外,其余部分均为合成;靶点上下游600bp序列为donorplasmid的左右同源臂序列信息,goi为avastin基因通过已有的诺维赞公司c115试剂盒完成抗体基因整合到donorplasmid上这个过程。
3、将cas9(丹麦科技大学dr.helenefkildegaard捐赠)、sgrna及donorplasmid3个质粒共同转染37℃、5%、co2条件下培养的cho细胞,三种质粒摩尔比为1:1:1,转染试剂为lipofectamine3000(thermofisherscientif-ic),具体转染方法参考说明书。之后细胞添加4μg/mlpuromycin进行筛选,过程共计10天。再用mofloxdpfacsmachine(beckmancoulter)进行单克隆细胞分选,选择不含任何荧光的细胞,接种到96孔板内。
4、细胞生长2周后,取一部分进行鉴定。通过做pcr鉴定,做5’junctionpcr,3’junctionpcr及out-outpcr进行鉴定。将阳性细胞保留。
测试例:
在对实施例4-6制得的这三个细胞系进行elisa检测,观察是否有目的蛋白质表达,且是否为稳定的长期表达。
检测的方法:三者的检测均通过elisa方法进行,在6孔板内培养挑选到的所有阳性细胞,检验其是否有目的蛋白质的长期稳定表达,使用的试剂盒为abclonal公司的humanalbuminelisakit(rk00157)和humanigg(total)eli-sakit(rk00393)进行实验。
图4与图5为hsa及nggh在不同传代下的细胞表达情况,纵坐标为每个细胞每天的分泌蛋白质质量。
可以看到图中nggh和hsa在平板内可以在50代以内稳定地表达相应基因,且挑选到的3个nggh定点整合细胞系及2个hsa定点整合细胞系目的蛋白质表达水平趋于接近。
图6为每日每个重组cho细胞分泌抗体蛋白的质量。显然,细胞在不同传代条件下均能稳定地表达分泌出相应的蛋白,体现了很好的稳定性,与之前的荧光细胞结果相互吻合。结果显示该位点可以进行crispr/cas9介导的定点整合,并可以稳定表达出相应的蛋白质。
在选取了5’-gaaagaaggtctgatatcaaagg-3’序列进行如上测试后,取得了良好结果,因此提出权利要求5-14所述靶序列皆能成功构建出定点整合稳定表达细胞系,且均能稳定表达出目的蛋白。
- 还没有人留言评论。精彩留言会获得点赞!