含吡啶并喹唑啉酮的三芳胺类化合物及其制备方法和应用与流程
本发明涉及含吡啶并喹唑啉酮的三芳胺类化合物及其制备方法和应用。
背景技术:
染料敏化太阳能电池(dssc)由于易制作、效率高和成本低,被认为是传统的昂贵和低效率的单晶硅太阳能电池的较好替代品。
在组成染料敏化太阳能电池的各个成分中,具有优良吸光性能和电子传输性能的染料敏化剂是保证dssc光电转换效率的最主要因素。国内外染料敏化剂的研究主要分两类,一类为有机金属配合物,典型结构为功能性多吡啶钌化合物,第二类为非金属有机染料。其中非金属有机染料具有较高的消光系数、简单的制备和纯化过程以及低成本等优点,较含金属有机染料受到更多关注。根据染料敏化剂中电子给体结构不同,已有大量非金属染料敏化剂得以报道,例如香豆素型、吲哚啉型、咔唑型、三芳胺型等非金属染料敏化剂。其中三芳胺类非金属染料敏化剂研究最广,并取得了较好的光电转换效率。
吡啶并喹唑啉酮作为一类重要的含氮化合物,广泛存在于天然产物中,具有良好的生物活性。另外,吡啶并喹唑啉酮也是一类典型的含氮刚性芳香杂环化合物,具有良好的空穴传输能力和独特的光电性质,但其在染料中的应用则鲜有报道。
技术实现要素:
针对现有技术存在的上述技术问题,本发明的目的在于提供含吡啶并喹唑啉酮的三芳胺类化合物及其制备方法和应用,含吡啶并喹唑啉酮的三芳胺类化合物可以作为染料敏化剂使用,组装成的染料敏化太阳能电池具有较好的光电转换效率,为染料敏化剂的筛选增添了新的可应用物质。
所述的含吡啶并喹唑啉酮的三芳胺类化合物,其特征在于其分子结构如式(wl4)、式(wl5)、式(wl6)或式(wl7)所示;
所述的含吡啶并喹唑啉酮的三芳胺类化合物的制备方法,其特征在于将式(ii-1)、式(ii-2)、式(ii-3)或式(ii-4)所示化合物和氰乙酸溶于溶剂中,加入碱性物质,在氮气气氛保护下,加热回流搅拌反应,反应结束后,反应液进行浓缩除去溶剂,所得浓缩物溶于洗脱剂后通过柱层析硅胶进行分离提纯,收集洗脱液并蒸除洗脱剂,即制得目标产物;
式(ii-1)、式(ii-2)、式(ii-3)和式(ii-4)所示化合物的结构式如下:
所述的含吡啶并喹唑啉酮的三芳胺类化合物的制备方法,其特征在于加热回流搅拌反应的时间为5-8h。
所述的含吡啶并喹唑啉酮的三芳胺类化合物的制备方法,其特征在于溶剂为由氯仿和乙腈组成的混合溶剂或由二氯甲烷与乙腈组成的混合溶剂。
所述的含吡啶并喹唑啉酮的三芳胺类化合物的制备方法,其特征在于洗脱剂为由二氯甲烷和甲醇组成的混合溶剂,所述二氯甲烷和甲醇的体积比为180~220:1,优选为200:1。
所述的含吡啶并喹唑啉酮的三芳胺类化合物的制备方法,其特征在于式(ii-1)、式(ii-2)、式(ii-3)或式(ii-4)所示化合物、氰乙酸和碱性物质的摩尔比为1:2-4:4-9,优选为1:3:4-9。
所述的含吡啶并喹唑啉酮的三芳胺类化合物的制备方法,其特征在于碱性物质为哌啶。
所述的含吡啶并喹唑啉酮的三芳胺类化合物的制备方法,其特征在于式(ii-1)、式(ii-2)、式(ii-3)或式(ii-4)所示化合物的物质的量与溶剂体积的比为1:20-140,物质的量的单位为mmol,体积的单位为ml。
所述的含吡啶并喹唑啉酮的三芳胺类化合物在染料敏化剂上的应用。
本发明以含吡啶并喹唑啉酮的三芳胺为给体,双键或噻吩作为桥键,氰乙酸作为受体,合成得到了含吡啶并喹唑啉酮的三芳胺类化合物,所述含吡啶并喹唑啉酮的三芳胺类化合物可作为染料敏化剂使用,该类化合物作为染料敏化剂组装成的染料敏化太阳能电池具有较好的光电转换效率,为染料敏化剂的筛选增添了新的可应用物质。
附图说明
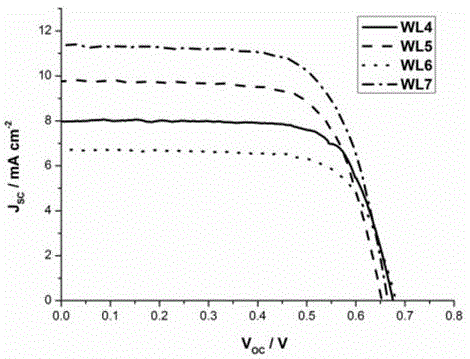
图1为本发明的含吡啶并喹唑啉酮的三芳胺类化合物组装得到的dssc的电流-电压曲线图。
具体实施方式
下面结合具体实施例对本发明作进一步说明,但本发明的保护范围并不限于此。
实施例1式(wl4)所示化合物的合成:
1)式(ii-1)所示化合物的合成
氮气保护下,向三口瓶中加入2-溴吡啶并喹唑啉酮(0.275g,1mmol)、联硼酸频那醇酯(0.253g,1mmol)、1,1'-双(二苯基膦)二茂铁氯化钯(分子式为pdcl2(dppf),0.022g,0.03mmol)、醋酸钾(0.294g,3mmol)和1,4-二氧六环(15ml),加热回流搅拌反应10h,反应结束后,向反应液中加入100ml水,再加入30ml二氯甲烷进行萃取,静置分层,分液得有机层和水层,有机层依次经饱和食盐水洗涤、无水na2so4干燥,再浓缩蒸除溶剂得到粗产物i,直接投下一步反应。
氮气保护下,粗产物i(0.352g,1mmol)、式(iii)所示化合物(0.322g,1mmol)、pd(pph3)4(0.15g,0.13mmol)、k2co3(0.40g,2.90mmol)溶于40ml溶剂中(溶剂为v甲醇:v水:v甲苯=1:2:37),加热回流搅拌反应10h,反应结束后,反应液蒸除溶剂,蒸除溶剂后的残余物用二氯甲烷溶解,再依次经饱和氯化钠溶液洗涤、无水na2so4干燥,然后浓缩蒸除二氯甲烷,浓缩物通过柱层析分离得到式(ii-1)所示化合物橘黄色粉末0.25g(收率54%),其中洗脱剂为体积比为1:10的石油醚和二氯甲烷,其中柱层析分离过程:浓缩物溶于洗脱剂后通过柱层析硅胶进行分离提纯,收集洗脱液并蒸除洗脱剂,干燥得到固体粉末产品,以下实施例等同。
式(ii-1)所示化合物:熔点253~256℃;1hnmr(500mhz,cdcl3)δ9.86(s,1h),8.93(d,j=7.3hz,1h),8.69(d,j=2.1hz,1h),8.14(dd,j=8.7,2.2hz,1h),7.89(d,j=8.7hz,1h),7.73(dt,j=8.6,6.1hz,4h),7.57(d,j=3.5hz,2h),7.40(t,j=8.5hz,2h),7.30(d,j=8.6hz,2h),7.27-7.20(m,3h),7.13(d,j=8.7hz,2h),6.94-6.91(m,1h).hrms(esi)m/z468.1707[m+h]+,calcdforc31h22n3o2:468.1712。
2)式(wl4)所示化合物的合成:
氮气保护下,将式(ii-1)所示化合物(0.1g,0.21mmol)、氰乙酸(0.053g,0.63mmol)及哌啶(0.1ml)溶于20ml溶剂中(溶剂为v三氯甲烷:v乙腈=1:2),加热回流搅拌反应8h,反应结束后,反应液浓缩后通过柱层析分离(洗脱剂为v二氯甲烷:v甲醇=300:1)得到式(wl4)所示化合物的红色粉末0.09g(收率80%)。
式(wl4)所示化合物:熔点260~262℃;
1hnmr(500mhz,dmso)δ8.84(d,j=7.3hz,1h),8.54(d,j=2.2hz,1h),8.27(dd,j=8.7,2.2hz,1h),8.15(s,1h),7.96(d,j=8.0hz,2h),7.88(d,j=8.6hz,2h),7.84(d,j=8.7hz,1h),7.76-7.73(m,1h),7.56(d,j=9.1hz,1h),7.47(t,j=8.0hz,2h),7.31(d,j=8.6hz,2h),7.29-7.26(m,3h),7.11-7.08(m,1h),7.00(d,j=8.9hz,2h).hrms(esi)m/z535.1761[m+h]+,calcdforc34h23n4o3:535.1770。
实施例1中合成式(wl4)所示化合物的反应路线如下:
实施例2式(wl5)所示化合物的合成:
1)式(ii-2)所示化合物的合成
氮气保护下,式(iv)所示化合物(0.434g,1mmol)、碳酸钾(0.276g,2mmol)、四三苯基膦钯(0.11g,0.1mmol)和式(i)所示化合物(0.32g,1mmol)溶于50ml溶剂中(溶剂为v甲苯:v甲醇:v水=6:1:2),加热回流搅拌反应10h,反应结束后,将反应液冷却至室温,过滤,滤液蒸除溶剂,蒸除溶剂后的残余物用二氯甲烷溶解,再依次经饱和氯化钠溶液洗涤、无水na2so4干燥,然后浓缩蒸除二氯甲烷,浓缩物通过柱层析分离得到式(ii-2)所示化合物橘黄色粉末0.32g(收率58%),其中洗脱剂为体积比为3:1的石油醚和乙酸乙酯。
式(ii-2)所示化合物:熔点265~268℃;1hnmr(500mhz,cdcl3)δ9.88(s,1h),8.98(d,j=7.1hz,1h),8.68(d,j=2.1hz,1h),8.16(dd,j=8.7,2.2hz,1h),7.99(d,j=8.6hz,1h),7.74(d,j=4.0hz,1h),7.69-7.67(m,3h),7.58(d,j=9.0hz,2h),7.38-7.35(m,3h),7.27-7.25(m,3h),7.22(d,j=7.6hz,2h),7.17-7.15(m,3h),7.02(t,j=7.1hz,1h).hrms(esi)m/z550.1581[m+h]+,calcdforc35h24n3o2s:550.1589
2)式(wl5)所示化合物的合成
氮气保护下,将式(ii-2)所示化合物(0.32g,0.58mmol)、氰乙酸(0.146g,1.74mmol)及哌啶(0.5ml)溶于15ml混合溶剂(混合溶剂为v三氯甲烷:v乙腈=1:2),加热回流搅拌反应8h,反应结束后,反应液浓缩后通过柱层析分离(洗脱剂为v二氯甲烷:v甲醇=200:1)得到式(wl5)所示化合物的红色粉末0.28g(收率80%)。
式(wl5)所示化合物:熔点>300℃;1hnmr(500mhz,dmso)δ8.84(d,j=7.3hz,1h),8.53(d,j=2.0hz,1h),8.26(dd,j=8.7,2.1hz,1h),8.21(s,1h),7.85-7.79(m,4h),7.76-7.72(m,1h),7.70(d,j=8.7hz,2h),7.59(d,j=3.8hz,1h),7.56(d,j=9.1hz,1h),7.41(t,j=7.8hz,2h),7.21-7.17(m,5h),7.11-7.09(m,3h).hrms(esi)m/z617.1647[m+h]+,calcdforc38h25n4o3s:617.1647。
实施例2中合成式(wl5)所示化合物的反应路线如下:
实施例3式(wl6)所示化合物的合成:
1)式(ii-3)所示化合物的合成
氮气保护下,向三口瓶中加入2-溴吡啶并喹唑啉酮(0.275g,1mmol)、联硼酸频那醇酯(0.253g,1mmol)、pdcl2(dppf)(0.022g,0.03mmol)、醋酸钾(0.294g,3mmol)和1,4-二氧六环(15ml),加热回流搅拌反应10h,反应结束后,向反应液中加入100ml水和30ml二氯甲烷,萃取,静置分层,分液得有机层和水层,有机层依次经饱和食盐水洗涤、无水na2so4干燥,再浓缩蒸除溶剂得到粗产物i,直接投下一步反应。
氮气保护下,粗产物i(0.148g,0.4mmol)、式(v)所示化合物(0.322g,1mmol)、pd(pph3)4(0.15g,0.13mmol)和k2co3(0.40g,2.90mmol)溶于50ml溶剂(溶剂为v甲苯:v甲醇:v水=37:1:2),加热回流搅拌反应16h,反应结束后,反应液蒸除溶剂,蒸除溶剂后的残余物用二氯甲烷溶解,再依次经饱和氯化钠溶液洗涤、无水na2so4干燥,然后浓缩蒸除二氯甲烷,浓缩物通过柱层析分离得到式(ii-3)所示化合物橘黄色粉末0.33g(收率50%),其中洗脱剂为二氯甲烷。
式(ii-3)所示化合物:1hnmr(500mhz,cdcl3)δ9.89(s,1h),8.94(d,j=7.4hz,2h),8.72(d,j=2.1hz,1h),8.16(dd,j=8.7,2.2hz,2h),7.90(d,j=8.7hz,2h),7.80-7.74(m,5h),7.58-7.54(m,5h),7.48(s,1h),7.37-7.35(m,3h),7.23(d,j=8.7hz,2h),7.06(s,1h),6.94-6.91(m,2h);hrms(esi)m/z662.2153[m+h]+,calcdforc43h28n5o3:662.2187。
2)式(wl6)所示化合物的合成
氮气保护下,将式(ii-3)所示化合物(0.1g,0.15mmol)、氰乙酸(0.0378g,0.45mmol)及哌啶(0.1ml)溶于20ml溶剂(溶剂为v三氯甲烷:v乙腈=1:2),加热回流搅拌反应8h,反应结束后,反应液浓缩后通过柱层析(洗脱液为v二氯甲烷:v甲醇=300:2)分离得到式(wl6)所示化合物的红色粉末0.82g(收率75%)。
式(wl6)所示化合物:熔点>300℃;1hnmr(500mhz,dmso)δ8.84(d,j=7.2hz,2h),8.54(d,j=2.1hz,2h),8.28(dd,j=8.8,2.0hz,2h),7.93(s,1h),7.91-7.80(m,8h),7.76-7.71(m,2h),7.56(d,j=9.2hz,2h),7.32-7.30(m,3h),7.20(d,j=8.7hz,1h),7.14(d,j=8.8hz,2h),7.10(t,j=6.9hz,2h).hrms(esi)m/z751.2069[m+na]+,calcdforc46h28n6nao4:751.2070。
实施例3中合成式(wl6)所示化合物的反应路线如下:
实施例4式(wl7)所示化合物的合成
1)式(ii-4)所示化合物
氮气保护下,式(vi)所示化合物(0.434g,1mmol)、碳酸钾(0.552g,4mmol)、四三苯基膦钯(0.33g,0.29mmol)和式(i)所示化合物(0.96g,3mmol)溶于50ml混合溶剂中(混合溶剂为v甲苯:v甲醇:v水=6:1:2),加热回流搅拌反应15h,反应结束后将反应液冷却至室温,过滤,滤液蒸除溶剂,蒸除溶剂后的残余物用二氯甲烷溶解,再依次经饱和氯化钠溶液洗涤、无水na2so4干燥,然后浓缩蒸除二氯甲烷,浓缩物通过柱层析分离得到式(ii-4)所示化合物橘黄色粉末0.35g(收率47%),其中洗脱剂为体积比为2:1的石油醚和乙酸乙酯。
式(ii-4)所示化合物:熔点>300℃;1hnmr(500mhz,cdcl3)δ9.89(s,1h),8.94(d,j=7.4hz,2h),8.71(d,j=2.1hz,2h),8.16(dd,j=8.7,2.2hz,2h),7.89(d,j=8.7hz,2h),7.76-7.73(m,5h),7.63(d,j=8.5hz,2h),7.57-7.56(m,3h),7.38(d,j=4.0hz,1h),7.33(d,j=8.6hz,4h),7.25(d,j=8.7hz,3h),6.96-6.90(m,2h).hrms(esi)m/z744.2057[m+h]+,calcdforc47h30n5o3s:744.2069。
2)式(wl7)所示化合物的合成
氮气保护下,将式(ii-4)所示化合物(0.35g,0.47mmol)、氰乙酸(0.118g,1.41mmol)及哌啶(0.4ml)溶于15ml混合溶剂(混合溶剂为v三氯甲烷:v乙腈=1:2),加热回流搅拌反应10h,反应结束后,反应液浓缩后通过柱层析分离(洗脱剂为v二氯甲烷:v甲醇=200:1)得到式(wl7)所示化合物的橘红色粉末0.27g(收率71%)。
式(wl7)所示化合物:熔点>300℃;1hnmr(500mhz,dmso)δ8.85(d,j=7.1hz,2h),8.55(d,j=2.2hz,2h),8.43(s,1h),8.29(dd,j=8.8,2.2hz,2h),7.98(d,j=3.5hz,1h),7.86(t,j=8.9hz,6h),7.79-7.76(m,2h),7.75-7.72(m,2h),7.69(d,j=4.0hz,1h),7.57(d,j=9.1hz,2h),7.30(d,j=8.6hz,4h),7.20(d,j=8.7hz,2h),7.10(t,j=6.9hz,2h).hrms(esi)m/z811.2129[m+h]+,calcdforc50h31n6o4s:811.2127。
实施例4中合成式(wl7)所示化合物的反应路线如下:
实施例5含吡啶并喹唑啉酮的三芳胺类化合物作为染料敏化剂的应用:
当作为染料敏化剂时,所述应用包括以下步骤:
将含吡啶并喹唑啉酮的三芳胺类化合物溶于ch3cl-ch3oh混合溶剂中,得到含吡啶并喹唑啉酮的三芳胺类化合物溶液(含吡啶并喹唑啉酮的三芳胺类化合物的浓度为3×10-4mol·l-1);所述ch3cl-ch3oh混合溶剂中ch3cl和ch3oh的体积比为10:1;
利用丝网印刷制备的双层tio2纳米粒子膜作为光电极:首先在导电玻璃fto上印一层12μm厚的20nm的tio2粒子,于450℃下马弗炉内煅烧30min,将煅烧后的膜冷却至室温后浸入0.04mol·l-1的ticl4水溶液70℃预处理30min,然后将膜从ticl4水溶液中取出并分别用水和乙醇冲洗,电吹风吹干。经马弗炉450℃下再次煅烧30min后,即得双层tio2纳米粒子膜光电极。
将上述煅烧得到的双层tio2纳米粒子膜光电极冷却至80℃后浸入上述得到的含吡啶并喹唑啉酮的三芳胺类化合物溶液中,室温敏化24h,得到负载含吡啶并喹唑啉酮的三芳胺类化合物的tio2电极。
铂对电极的制备:采用丝网印刷方法,将h2ptcl6水溶液印刷在fto导电玻璃上,h2ptcl6水溶液将fto导电玻璃表面润湿,干燥,然后在马弗炉400℃烧结20min,即制得铂对电极。
将上述制得的双层tio2纳米粒子膜光电极和铂对电极组装成三明治结构,在所述三明治结构边缘滴入电解质,利用毛细管渗透原理引入电池内部,组装形成染料敏化太阳能电池(dssc),于100mw/cm2光强照射下,测定含吡啶并喹唑啉酮的三芳胺类化合物组装得到的dssc的电流-电压曲线如图1所示,其性能参数结果如表1所示:
表1含吡啶并喹唑啉酮的三芳胺组装得到的dssc性能参数
从表1可以看出:本发明含吡啶并喹唑啉酮类三芳胺类化合物作为染料敏化剂组装成的染料敏化太阳能电池具有较好的光电转换效率,光电转换效率为3.50~5.19%,为染料敏化剂的筛选增添了新的可应用物质。
本说明书所述的内容仅仅是对发明构思实现形式的列举,本发明的保护范围不应当被视为仅限于实施例所陈述的具体形式,本发明的保护范围也仅仅于本领域技术人员根据本发明构思所能够想到的等同技术手段。
- 还没有人留言评论。精彩留言会获得点赞!