曲索芬头孢曲松钠药物制剂治疗细菌性内膜炎的新适应症的制作方法
本发明涉及药物制备技术,具体涉及一种头孢曲松钠组合物、制备方法及其应用。
背景技术:
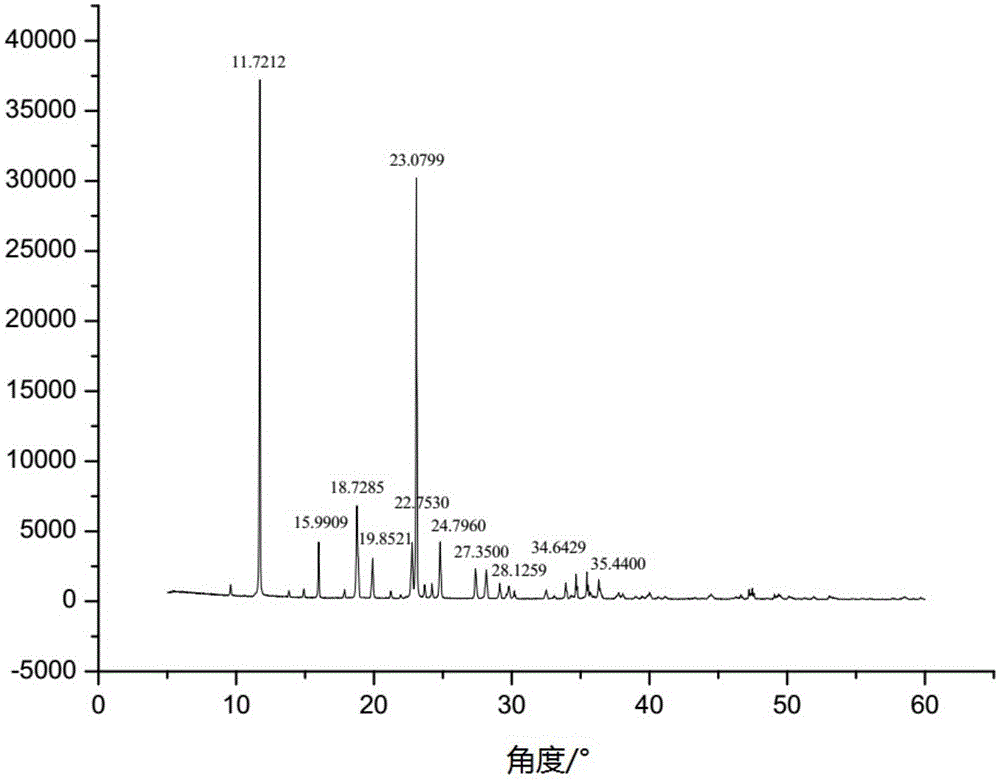
:头孢曲松钠(ceftriaxonesodium)是第三代头孢菌素类抗生素。由罗氏公司原研上市,商品名为罗氏芬(rocephin),国际公认的品牌有:曲索芬、菌必治、acantex、cefaxone。化学名称是(6r,7r)-7-[[(2-氨基-4-噻唑基)(甲氧亚氨基)乙酰]氨基]-8-氧代-3-[[(1,2,5,6-四氢-2-甲基-5,6-二氧代-1,2,4-三嗪-3-基)硫代]甲基]-5-硫代-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸二钠盐三倍半水合物。其化学结构式如下:在药物使用中可能会出现关于药物的不良反应的可能,常见因素包括剂量、给药方案、疗程、人口统计学特征、联合用药、效能特征等,但与用药不良反应重要相关且可控的因素为药物杂质。虽然药典中规定了药品的含量、有关物质及其他杂质(水分、重金属、可见异物、不溶性微粒、细菌内毒素、溶剂等)的限量,但大量的研究均表明了随着药品含量或纯度的提升,对药品的安全性和疗效起到促进作用。因此,控制杂质水平在药物开发研究过程中越来越受到医药工作者和医药企业的重视。我们对头孢曲松钠的稳定性进行了研究,发现头孢曲松钠对强酸、强碱、氧化和温度均不稳定,加之原料杂质和生产过程中产生的杂质在成品中的残留,使得成品制剂在储存过程中的稳定性进一步弱化。而目前头孢曲松钠的单方制剂和复方制剂的生产工艺或储存方法均未能提出行之有效的解决方案。目前大量的研究已经证实,药物中的杂质多数具有潜在的生物活性,影响药物的安全性和有效性,甚至产生毒性。虽然尚未有研究能够充分证实这些杂质对人体造成的伤害,但其毕竟是药品中的污染物,不具有治疗作用,应尽可能将其降到最低水平。我们知道,有机药物晶体大多是分子晶体,可因结晶条件不同而得到不同的晶型,这种现象称为多晶型。药物的晶型不同,他们的物理性质如密度、熔点、硬度、溶解度等方面均有显著差异。药物的多晶型也对药品质量及临床疗效也有较大的影响,研究药物的多晶型有利于改造药物,减缓储藏时药物的降解,减轻毒副反应。目前,晶型的研究越来越受到关注,但能够有效展现出利于储藏稳定性的头孢曲松钠晶体的制备方法仍然较少。基于头孢曲松钠杂质对用药效果和安全性的考虑,有必要对头孢曲松钠制剂生产工艺和制剂形式进行研究,以解决头孢曲松钠储藏时稳定性不足,以及杂质带来的头孢曲松钠制剂稳定性差、安全性隐患的问题。技术实现要素:本发明人经过锐意研究,通过对原料合成工艺进行改进,提供了一种低杂质含量的头孢曲松钠新晶型化合物,该新晶型化合物储藏稳定性较现有晶型化合物高;制备该新晶型化合物的工艺能够有效控制产品中的杂质,有利于提高产品或相应制剂的使用安全性和相应的临床治疗效果,从而完成本发明。本发明目的是提供以下技术方案:(1),头孢曲松钠或其化合物,其中有效组分头孢曲松钠的质量含量大于98%,头孢曲松钠或其化合物中还包括,杂质a为其质量含量不高于0.2%;优选地,杂质b为其质量含量不高于0.2%;杂质c为头孢曲松钠的反式异构体其质量含量不高于0.2%。优选地,其组分头孢曲松钠的x-射线粉末衍射图谱中特征峰的2θ值包括11.72±0.2°,15.99±0.2°,18.72±0.2°,19.85±0.2°,22.75±0.2°,23.08±0.2°,24.79±0.2°,27.35±0.2°,28.12±0.2°,34.64±0.2°,35.44±0.2°,其中,“±0.2”为允许的测量误差范围。(2)一种权利要求(1)所述的头孢曲松钠或其组合物中头孢曲松钠的制备方法,该制备方法以下步骤:步骤1),7-氨基头孢烷酸(7-aca)和2,5-二氢-6-羟基-2-甲基-3-巯基-5-氧-1,2,4-三嗪(ttz)在催化剂作用下,合成化合物7-act(7-氨基-3-[(2,5-二氢-6-羟基-2-甲基-5-氧代-1,2,4-三嗪-3-巯基)甲基]-3-头孢-4-羧酸);步骤2),7-act与ae-活性酯反应,反应完成后加入成盐剂,合成头孢曲松钠粗品;步骤3),对头孢曲松钠粗品进行纯化,干燥,得到高纯度头孢曲松钠水合物;优选地,纯化包括重结晶;更优选地,结晶溶剂为有机溶剂-水溶液,优选为乙醇水溶液或丙二醇水溶液,更优选为乙醇体积浓度为15%~25%的水溶液;结晶溶剂的体积与头孢曲松钠粗品的质量之比为(1.5~2.0):1;析晶溶剂为乙醇、丙二醇中的一种或多种,优选为乙醇;结晶过程中析晶溶剂的体积与头孢曲松钠粗品的质量之比为(1.5~2.0):1。(3)一种头孢曲松钠的单方制剂,其包括以上述(1)所述的头孢曲松钠或其组合物、或上述(2)所述的制备方法制得的头孢曲松钠为活性成分,所述制剂类型包括注射剂、颗粒剂、片剂、滴丸剂、胶囊剂,优选为注射剂。(4)一种头孢曲松钠的复方制剂,其选用上述(1)所述的头孢曲松钠或其组合物、或上述(2)所述的制备方法制得的头孢曲松钠为活性成分,所述复方制剂由包括以下质量配比的原料成分制成:头孢曲松钠10份;增效剂1~10份;其中,所述增效剂为阿维巴坦、舒巴坦、他唑巴坦或其钠盐;任选地,所述复方制剂中还可以包括药学上可接受的没有配伍禁忌的辅料和/或药学活性物质,所述辅料优选为渗透压调节剂、ph调节剂和抗氧化剂中的一种或多种。(5)上述(1)所述的头孢曲松钠组合物、上述(2)所述的制备方法制得的头孢曲松钠、上述(3)所述的头孢曲松钠的单方制剂、或上述(4)所述的头孢曲松钠的复方制剂,在制备治疗细菌性内膜炎的药物方面的用途。根据本发明提供的一种头孢曲松钠或其组合物、制备方法及其应用,具有以下有益效果:(1)根据本发明提供的头孢曲松钠的制备方法,有效保证了原料高含量、低杂质,有利于提高使用安全性和相应制剂的临床治疗效果;(2)本发明方法制备得到的头孢曲松钠组合物为新晶型,该晶型储藏稳定性和溶解性突出,引湿性低,便于长期存储和放置,减少了降解产生杂质的量,从而降低临床致敏风险;(3)根据本发明提供的头孢曲松钠组合物及其制剂,具有有效的制备治疗细菌性内膜炎的药物方面的应用。附图说明图1示出实施例1-1中制备得到的头孢曲松钠晶体的xrd图谱;图2示出头孢曲松钠标准品(中国药品生物制品检定所,批号:130480-201504)的xrd图谱;图3示出实施例1-1中制备得到的头孢曲松钠晶体的dsc图谱;图4示出患有细菌性内膜炎患者的头孢曲松钠药时曲线;图5示出患有细菌性内膜炎患者的舒巴坦钠药时曲线。具体实施方式以下通过具体实施方式本发明进行进一步说明,本发明的特点和优点将随着这些说明而变得更为清楚。本发明的目的在于提供一种头孢曲松钠或其组合物,本发明人对头孢曲松钠的合成工艺进行了研究,以期通过获得低相关杂质含量的头孢曲松钠,提高头孢曲松钠或其组合物及其相关制剂的稳定性和用药安全性。在一种优选的实施方式中,本发明提供的头孢曲松钠或其组合物中,有效组分头孢曲松钠(c18h16n8na2o7s3·3.5h2o)的质量含量大于98%,甚至大于99%。在一种优选的实施方式中,本发明中提供的头孢曲松钠的x-射线粉末衍射图谱中特征峰的2θ值包括11.72±0.2°,15.99±0.2°,18.72±0.2°,19.85±0.2°,22.75±0.2°,23.08±0.2°,24.79±0.2°,27.35±0.2°,28.12±0.2°,34.64±0.2°,35.44±0.2°,其中,“±0.2”为允许的测量误差范围。本发明人进行了大量的试验和检索,发现本发明提供的头孢曲松钠为不同于现有技术中头孢曲松钠晶体的新晶型。文献“薛晶,贾燕花,李进,尹利辉,胡昌勤.头孢曲松钠的亚晶型分类及对产品质量的影响[j].药学学报,2014,49(07):1034-1038.”示出的头孢曲松钠样品的xrd图谱中,在2θ为11.1°,12.4°,15.4°,18.3°,18.8°,19.9°,21.1°,22.6°,23.7°,24.5°,25.2°,26.8°,27.7°,28.2°,29.5°,30.7°,33.7°,36.4°,37.8°,41.6°,43.1°,44.4°处存在特征峰,与标准品xrd谱图吻合。很明显,本发明中头孢曲松钠xrd的特征峰与该文献和标准品不一致。在本发明中,头孢曲松钠的合成工艺包括以下步骤:步骤1),7-氨基头孢烷酸(7-aca)和2,5-二氢-6-羟基-2-甲基-3-巯基-5-氧-1,2,4-三嗪(ttz)在催化剂作用下,合成化合物7-act(7-氨基-3-[(2,5-二氢-6-羟基-2-甲基-5-氧代-1,2,4-三嗪-3-巯基)甲基]-3-头孢-4-羧酸);步骤2),7-act与ae-活性酯(2-(2-氨基-4-噻唑基)-2-(甲氧亚氨基)乙酸硫代苯并噻唑酯)反应,反应完成后加入成盐剂,得到头孢曲松钠粗品;步骤3),对头孢曲松钠粗品进行纯化,干燥,得到高纯度头孢曲松钠水合物。步骤1),将7-aca溶于反应溶剂后,加入ttz和催化剂进行反应,合成化合物7-act。反应如下式(1)所示:由合成路线可知,反应的起始原料7-aca和ttz都有可能出现在最终产品中,ttz的毒性尚无定论,但7-aca已经证实对豚鼠具有较强至敏性,对人体产生至敏性的可能性极大。同时,反应过程中,若反应速度慢且7-aca过量较多,容易导致7-aca自缩合(氨基和酯基反应),产生高分子化合物的可能性,而高分子化合物为引起过敏反应的主要原因之一,故而7-aca反应程度和用量关系到后处理难度和产品的安全性。本发明人进行了大量研究,选择ttz微过量,结合反应条件如溶剂和催化剂的设定,能够实现反应产物的高效转化,降低反应原料在产物中的残存量。具体地,确定7-氨基头孢烷酸(7-aca)和2,5-二氢-6-羟基-2-甲基-3-巯基-5-氧-1,2,4-三嗪(ttz)的摩尔比为1:(1.05~1.20),优选为1:(1.10~1.15)。在步骤1)中,利用弱酸催化剂对反应进行催化,本发明人选择三氯化铝(alcl3)-三氟化硼(bf3)碳酸二甲酯复合催化剂。经过大量实验研究,本发明人发现,相较于传统的三氟化硼乙腈络合物或者三氟化硼乙醚络合物,alcl3-bf3碳酸二甲酯复合催化剂具有更高的催化效率,主要表现在对反应原料的高转化率、高反应速率,这相当于提高了反应原料稳定性(避免7-aca自缩合产生高分子化合物杂质),降低了反应原料的剩余。优选地,复合催化剂中alcl3和bf3的用量以两者的质量比计,alcl3和bf3的摩尔比为(0.125~0.2):1,bf3在催化剂中的质量含量约为18%~20%。复合催化剂的总用量以复合催化剂中bf3与7-aca的用量比计,bf3与7-aca的质量比为1:(20~50)。催化剂以滴加的方式加入,滴加过程中反应体系温度控制在5℃~10℃,滴加完成后升温至20~30℃保温0.5~1.5h。在步骤1)中,反应溶剂为甲苯、邻二甲苯、对二甲苯、乙腈、二氯甲烷或碳酸二甲酯中的任意一种或多种,优选为乙腈/二氯甲烷(v乙腈/v二氯甲烷=5:1)或碳酸二甲酯,更优选为碳酸二甲酯。反应物浓度与反应速率直接相关,为促进反应进行及便于搅拌(涉及反应均匀性),7-aca与反应溶剂的用量比以7-aca的质量与反应溶剂的体积之比计为1:(3~5),以1g为1质量份,1ml为1体积份。在步骤1)中,7-aca和ttz反应完全后,向反应体系中加入水,优选还加入抗氧化剂,加入过程中伴随搅拌,缓慢滴加氨水调节体系ph至2.5,降温至10~15℃、养晶,抽滤、干燥,得化合物7-act。优选地,所述抗氧化剂为2%连二亚硫酸钠溶液。步骤2),7-act与ae-活性酯反应,反应完成后加入成盐剂,合成头孢曲松钠。反应如下式(2)所示:步骤2)包括以下子步骤:子步骤2-1),将7-act、ae-活性酯溶于反应溶剂中,降温并控制温度为0℃~5℃,加入催化剂,保温反应2h~4h,优选为3h;子步骤2-2),反应完成后,加入成盐剂,升温至20℃~30℃,加入析晶溶剂,滴加完毕后降温至5℃~10℃,搅拌,析晶,抽滤,干燥,得到头孢曲松钠粗品。在步骤2-1)中,7-act和ae-活性酯的用量以其摩尔比计为1:(1.01~1.20),优选为1:(1.05~1.10)。7-act会诱发豚鼠的躁动、颤抖、喷嚏、骚鼻等过敏反应症状,而过量的ae-活性酯则会在后续反应中分解或利于通过结晶去除,因而在药品中极少残留,故而选择ae-活性酯微过量,结合催化剂和反应条件,促进原料转化。在步骤2-1)中,所述反应溶剂为甲苯、邻二甲苯、对二甲苯、乙腈或二氯甲烷中的任意一种或多种,优选为乙腈/二氯甲烷(v乙腈/v二氯甲烷=5:1)混合溶剂。在步骤2-1)中,所述催化剂为有机胺,如脂肪胺类、醇胺类、脂环胺类、芳香胺类,优选为脂肪胺类,更优选为吡啶、三乙胺、乙二胺、异丙胺、二异丙胺、正丁胺、异丁胺、1,4-丁二胺、三正丁基胺、己胺和己二胺中一种或多种,最优选为三正丁基胺和吡啶的混合物。现有技术中,通常采用三乙胺作为催化剂,然而本发明人发现,当采用三乙胺作为催化剂时结晶得到的头孢曲松钠粗品常常为黄色,增加后续精制的难度,通常需要两次以上的再结晶得到颜色合格的产品,增加了生产流程,影响了产品的收率。本发明人对此进行了大量研究,发现选用三正丁基胺和吡啶的混合物作为催化剂获得的头孢曲松钠粗品颜色相较于三乙胺催化时明显减轻。值得注意的是,催化剂的调整,促进了反应速率的提升,相应的降低了生产过程中副反应的发生。特别地,催化剂与7-act的摩尔比为1:(1.0~3.0),优选为1:(1.0~1.5)。催化剂选用三正丁基胺和吡啶的混合物时,三正丁基胺和吡啶的摩尔比为(5~10):1。在子步骤2-2)中,所述成盐剂选自碳酸氢钠、醋酸钠或异辛酸钠中的一种或多种,优选为异辛酸钠。在子步骤2-2)中,所述析晶溶剂为丙酮、乙醇、甲醇中的一种或多种,优选为丙酮。由式(2)可知,该步骤反应会相应生成2-巯基苯并噻唑,经过研究发现,2-巯基苯并噻唑会对豚鼠产生过敏反应,如躁动、颤抖等。然而,2-巯基苯并噻唑的产生不可避免,需要采用有效手段将其与产品分离。较为常用的手段为结晶法分离产品和杂质,此时反应溶剂和析晶溶剂的组合对粗产品中夹带杂质有重要影响,本发明人对此进行了大量研究,惊奇地发现,在子步骤2-1)中反应溶剂为乙腈/二氯甲烷(v乙腈/v二氯甲烷=5:1)混合溶剂、且子步骤2-2)中析晶溶剂为丙酮时,相较于其他反应溶剂和析晶溶剂的组合,能够更加高效地去除反应原料和副产物如2-巯基苯并噻唑在头孢曲松钠粗品中的残留,主要体现在最终产品有效含量(或杂质含量)上。步骤3),对头孢曲松钠粗品的纯化,干燥,得到高纯度头孢曲松钠水合物;具体地,将头孢曲松钠粗品溶解于结晶溶剂中,加入抗氧剂,活性炭脱色,微孔滤膜过滤,滴加析晶溶剂,梯度降温析晶,抽滤,干燥,得到高纯度头孢曲松钠水合物。步骤3)中,所述抗氧化剂选自硫酸氢钠或者连二亚硫酸钠。步骤3)中,所述结晶溶剂为有机溶剂-水溶液,优选为乙醇水溶液或丙二醇水溶液,更优选为乙醇体积浓度为15%~25%的水溶液。结晶溶剂的体积与头孢曲松钠粗品的质量之比为(1.5~2.0):1,其中,以1g为1重量份,1ml为1体积份。本发明中上述结晶溶剂能够保证头孢曲松钠粗品和杂质的溶解且有机溶剂用量少。步骤3)中,所述析晶溶剂为乙醇、丙二醇中的一种或多种,优选为乙醇。特别地,结晶过程中析晶溶剂的体积与头孢曲松钠粗品的质量之比为(1.5~2.0):1,其中,以1g为1重量份,1ml为1体积份。步骤3)中,降温过程中不断搅拌,搅拌速率为80~100转/分钟。特别地,本发明采用梯度降温析晶方法。析晶过程为:第一阶段:先加入一半的析晶溶剂,搅拌速率为80~100转/分钟,降温至10~15℃,保温0.5~1.5h;第二阶段:再降温至5~8℃,保温1.5~2.5h;第三阶段:保温结束后,继续滴加另一半析晶溶剂,滴加时间为1.5-2.0h,滴加完毕后搅拌1.5~2.5h析晶。本发明中,在第一阶段,随着温度降低和析晶溶剂的加入,头孢曲松钠在结晶溶剂中达到饱和;在第二阶段,温度继续降低使得头孢曲松钠在结晶溶剂中达到过饱和,逐渐析晶;在第三阶段,再次滴加析晶溶剂,在低温下进一步搅拌析晶,获得纯化后产物。其中,第一阶段中保温的目的在于加入一半析晶溶剂后,降温,搅拌保温,促进溶液生成过饱和溶液,但是未有晶体析出;第二阶段中保温的目的在于析出的晶体进行缓慢的生长,生成新的晶型,第三阶段中保温的目的在于进一步析晶,且为晶体的生长和晶型的调整提供足够的时间。优选地,第二阶段保温前,加入30~60目氯化钠,更优选氯化钠与结晶溶剂的用量比以氯化钠的质量与结晶溶剂的体积比计为(0.75~1.25):600,其中,以1g为1重量份,1ml为1体积份。氯化钠加入的目的在于使结晶溶剂中局部浓度过饱和,降低头孢曲松钠的溶解度,便于产物析出。选择氯化钠的粒径要求为30~60目,原因主要在于,较低目数的氯化钠在加入结晶体系的时候,会起到晶核的作用,诱导结晶的形成。我们知道,析晶过程主要由形成过饱和度—生成晶核—晶核的生长三步组成。其中,生成晶核的过程是控制产品晶型的关键。结晶温度、析晶溶剂的加入速度、过饱和度控制对晶核的生成以及最终产品的晶型有重要影响。对于结晶温度来说,头孢曲松钠的溶解度随温度的升高而升高,在结晶过程中,饱和度也随之变化,提高温度,有助于晶核的形成,有利于结晶过程;降低温度析晶、养晶,可以提高产品的收率,但温度过低,导致产品粒度过小,影响后续的分离、干燥。本发明人经过研究,过饱和度的形成采用梯度变温,第一阶段高温(10~15℃)有效溶解头孢曲松钠粗品,第二阶段降温为析晶做准备,第三阶段析晶温度(5~8℃)与第二阶段温度相同,仅通过加入析晶溶剂控制晶核的生成,变量条件少,通过控制了头孢曲松钠浓度(第一阶段和第二阶段)和过饱和度的稳定性(第二阶段和第三阶段),实现了晶核生成的可控性;例如,降低了析晶溶剂加入点处过饱和度水平很高、容易在瞬间产生大量晶核并形成聚集体(暴晶现象)、进而发生母液包藏、影响晶体产品质量的情况。同时,第三阶段析晶温度为5~8℃,不会由于温度过低造成产品粒径小,进而影响晶体的流动性和干燥效率的情况。对于析晶溶剂的加入速度来说,在没有晶体存在下(自发成核),晶核的产生对过饱和度的变化尤其敏感,析晶溶剂的加入速度过快则产生大量晶核并易形成聚集体;析晶溶剂的加入速度过慢则晶体粒径过大,且影响结晶效率。本发明中第三阶段,在设定头孢曲松钠浓度和析晶溶剂用量的前提下,确定了析晶溶剂的滴加时间(1.5-2.0h),同时加入氯化钠颗粒,避免了晶核的聚集和低生成效率。值得注意的是,本发明析晶溶剂的选择同样有助于避免暴晶现象。现有技术中普遍采用丙酮作为析晶溶剂,出现暴晶现象的概率并不低,然而在析晶溶剂的改善方面并未进行较多研究。本发明采用乙醇作为析晶溶剂,一方面与结晶溶剂中有机成分相同,便于回收,另一方面在于能够抑制暴晶现象产生,尤其与氯化钠协同作用下,能够实现晶核稳定产生和生长。进一步地,本发明在析晶体系中加入的氯化钠颗粒,还可以有效改善晶体的形状,相当于还起到了提高结晶体粒度和均一性的目的。选择30~60目的氯化钠,加入到体系中时,溶液为过饱和状态,氯化钠的加入,会作为晶种引导结晶的形成,形成的结晶重新作为晶种引导结晶,氯化钠会溶解于反应体系中。在上述条件范围内得到的头孢曲松钠晶体介于30~60μm,且粒度均匀性高,不会由于晶体颗粒过小产生易团聚、流动性差、混合均匀困难、稳定性较低的问题,也不会由于晶体颗粒较多大于100μm而导致装瓶后瓶间差异性大的问题。在本发明一种优选的实施方式中,活性炭的用量为头孢曲松钠粗品的1.0(质量)%~2.0(质量)%。活性炭的用量少,对有色杂质的去除效果不理想,然而,活性炭的用量上限也需严格控制,其对大分子物质的吸附容易将产物脱离结晶体系,影响产品收率。本发明人对产品进行分析时发现,头孢曲松(钠)在7位侧链酰胺键上脸有z型甲氧亚胺基,该基团在光照条件下容易转变成e型,形成头孢曲松(钠)反式异构体,如下式(3)。为此,在反应过程中,尤其是在结晶过程中,为防止头孢曲松反式异构体的生成,采取了避光操作。通过实验发现,避光条件下,杂质a生成的量远小于质控限度。避光操作主要体现在采用避光反应容装置上,如采用棕色避光玻璃反应装置进行合成或结晶。本发明人惊奇的发现,在上述结晶条件下获得的晶型不同于现有技术中公开的晶型,其x-射线粉末衍射图谱中特征峰的2θ值包括11.72±0.2°,15.99±0.2°,18.72±0.2°,19.85±0.2°,22.75±0.2°,23.08±0.2°,24.79±0.2°,27.35±0.2°,28.12±0.2°,34.64±0.2°,35.44±0.2°。我们知道,有机药物晶体可以因结晶条件不同而得到不同的晶型。药物的晶型不同,对药品质量有较大的影响,他们的物理性质如密度、熔点、硬度、溶解度等方面均有显著差异。本发明人对制备得到的头孢曲松钠晶体的稳定性和引湿性进行了评价,发现相较于市面上常见的头孢曲松钠注射针剂,本发明中头孢曲松钠晶体具有更加突出的稳定性,而引湿性则降低。本发明人对通过上述方法制备得到的头孢曲松钠进行了杂质分析,杂质a为头孢曲松钠的起始原料7-aca,其质量含量不高于0.2%,或者不高于0.1%,或者不高于0.05%,或者不高于0.01%;杂质b为2-巯基苯并噻唑,其质量含量不高于0.2%,或者不高于0.1%,或者不高于0.05%,或者不高于0.01%;杂质c为头孢曲松钠的反式异构体,其质量含量不高于0.2%,或者不高于0.1%,或者不高于0.05%,或者不高于0.01%。本发明中,杂质a主要是反应原料剩余产生的。一方面,我们通过ttz用量过量、以及对催化剂的特定选择,提高7-aca转化率,另一方面,优选乙腈/二氯甲烷(v乙腈/v二氯甲烷=5:1)或碳酸二甲酯为反应溶剂,并配合后续结晶过程中结晶溶剂和析晶溶剂的选择,可有效的去除杂质a。本发明中,杂质b主要是7-act与ae-活性酯反应生成头孢曲松时的副产物。本发明通过在该步骤中,采用反应溶剂为乙腈/二氯甲烷(v乙腈/v二氯甲烷=5:1)混合溶剂、析晶溶剂为丙酮,以及后续精制步骤对杂质b进行去除。本发明中,杂质c为反应过程中关照条件下生成的,本发明通过选用避光反应装置,使杂质a生成的量远小于质控限度。本发明中,采用上述方法得到头孢曲松钠晶体用于制备单方制剂,其可以是医药上可接受的任何制剂类型,包括注射剂、颗粒剂、片剂、滴丸剂、胶囊剂等或者负载在其他医学上可用的载体,优选为注射剂。本发明中,采用上述方法得到头孢曲松钠还可以与增效剂联用制备复方制剂,其可以是医药上可接受的任何制剂类型,包括注射剂、片剂、胶囊剂等或者负载在其他医学上可用的载体,优选为注射剂。在一种优选的实施方式中,所述复方制剂由包括以下质量配比的原料成分制成:头孢曲松钠10份;增效剂1~10份。其中,所述增效剂为阿维巴坦、舒巴坦、他唑巴坦或其钠盐。优选地,所述复方制剂中还可以包括药学上没有配伍禁忌的辅料和/或药学活性物质,所述辅料优选为渗透压调节剂、ph调节剂和抗氧化剂中的一种或多种。所述渗透压调节剂为氯化钠或葡萄糖;所述ph调节剂包括氢氧化钠、氯化钠、磷酸、磷酸二氢钠、磷酸氢二钠、醋酸钠中的一种或多种;所述抗氧化剂为维生素c、亚硫酸钠、焦亚硫酸钠中的任意一种或多种。本发明中,头孢曲松钠复方制剂通过混晶法得到,包括以下步骤:按配方精密称取头孢曲松钠和辅料,混合均匀,得到头孢曲松钠混合物,按配方精密称取增效剂,置于球磨机中研磨混合后,按设定规格依次向瓶中分装,密封即得。此时,头孢曲松钠、增效剂以及辅料的d90介于20~60μm,进一步为25~55μm。根据本发明提供的头孢曲松钠(或组合物)及其制备方法、头孢曲松钠的单方制剂或复方制剂,提供了一种用于制备治疗细菌性内膜炎的药物方面的用途。实施例以下通过具体优选的实施例对本发明进行进一步说明。这些实施例仅是说明性的,并不应理解为对本发明的限制。实施例1-1头孢曲松钠的合成步骤1),向反应瓶中加入7-aca272g(1.0mol),碳酸二甲酯1090ml,ttz175g(1.10mol),开启搅拌,反应体系降温至5℃~10℃,向反应体系中滴加9.4galcl3-bf3-碳酸二甲酯溶液[(wbf3)=18%](其中,alcl30.66g(0.005mol),bf31.7g(0.025mol)),滴加入完毕后,升温至20~30℃保温1h,再加入纯化水1500ml,滴加135ml2%的连二亚硫酸钠溶液,搅拌1h,用氨水调节ph至2.5,降温至10℃,抽滤,滤饼用乙腈/纯化水=1:3的混合液100ml淋洗两次,干燥得到7-act347.9g,收率:92%,纯度98.2%。步骤2),向反应瓶中加入步骤1)制得的7-act185.7g(0.50mol),ae-活性酯184g(0.525mol),乙腈750ml,二氯甲烷150ml,降温至0~5℃,加入94.5g三正丁基胺(0.51mol)和4.7g吡啶(0.06mol),温度不超过5℃,滴加完毕后保温3h,反应结束后,滴加配制好的异辛酸钠溶液700ml(含异辛酸钠620g),升温至约25℃,滴加丙酮5500ml,滴加时间为1h,滴加完毕后降温至5℃~10℃,搅拌0.5h,抽滤,干燥,得到白色头孢曲松钠粗品(含结晶水)为337g。步骤3),向反应瓶中加入步骤2)制得的头孢曲松钠粗品(含结晶水)200g,溶解于300ml注射用水和乙醇的混合溶剂(乙醇:水=1:5)中,加入抗氧剂连二亚硫酸钠,20~30℃下搅拌20min至固体溶清,再加入3g药用级别的针用活性炭脱色1h,0.2μm的微孔滤膜过滤去除活性炭,用15ml注射用水淋洗滤饼,合并滤液。滴加析晶溶剂乙醇150ml,滴加完毕后,控制转速在80~100转/分钟,降温至15℃,保温1h,然后缓慢降温至5~8℃,加入30~60目氯化钠0.5g,加入后保温2h,保温结束后继续滴加乙醇150ml,滴加时间为2.0h,滴加完毕后搅拌2h析晶,析晶结束后抽滤,用乙醇30ml淋洗滤饼两次,真空干燥得到头孢曲松钠水合物,189g,纯度为99.9%,收率94.4%,d9030~60μm。反应过程中全程避光操作。实施例1-2头孢曲松钠的合成与实施例1-1的合成工艺相同,区别仅在于:步骤1)中,向反应体系中滴加15galcl3-bf3-碳酸二甲酯溶液[(wbf3)=18%](其中,alcl30.66g(0.005mol),bf32.7g(0.04mol)),即alcl3-bf3碳酸二甲酯复合催化剂中alcl3和bf3的摩尔比为1:8。实施例1-3头孢曲松钠的合成与实施例1-1的合成工艺相同,区别仅在于:步骤2)中,反应溶剂由v乙腈:v二氯甲烷=5:1的混合溶剂,变更为乙腈,析晶溶剂为丙酮。实施例1-4头孢曲松钠的合成与实施例1-1的合成工艺相同,区别仅在于:步骤2)中,ae-活性酯192.7g(0.55mol),即7-act和ae-活性酯的质量比为1:1.0。实施例1-5头孢曲松钠的合成与实施例1-1的合成工艺相同,区别仅在于:步骤2)中,催化剂由三正丁基胺和吡啶,变更为相同摩尔量的三乙胺。得到头孢曲松钠粗品颜色为浅黄色。实施例1-6头孢曲松钠的合成与实施例1-1的合成工艺相同,区别仅在于:步骤3)中,不加入氯化钠颗粒。实施例2-1头孢曲松钠-舒巴坦钠(2:1)粉针剂所用原料如下:头孢曲松钠晶体其中,头孢曲松重量300g舒巴坦钠其中,舒巴坦重量为150g150瓶,3g/瓶(有效成分头孢曲松+舒巴坦=3g/瓶)采用以下生产工艺制备头孢曲松钠-舒巴坦钠粉针剂:按配方精密称取头孢曲松钠,按配方精密称取舒巴坦钠,并将原料均置于球磨机中研磨混合,以头孢曲松计2g装量,密封,包装,得到头孢曲松钠制剂。实施例2-2头孢曲松钠-他唑巴坦钠(3:1)粉针剂所用原料如下:头孢曲松钠晶体其中,头孢曲松重量300g他唑巴坦钠其中,他唑巴坦重量为100g200瓶,2g/瓶(有效成分头孢曲松+他唑巴坦=2g/瓶)头孢曲松钠-他唑巴坦钠粉针剂的制备方法同实施例2-1。对比例对比例1-1头孢曲松钠的合成与实施例1-1的合成工艺相同,区别仅在于:步骤1)中,将alcl3-bf3碳酸二甲酯复合催化剂替换为bf3乙腈催化剂。得到7-act330g,收率:86.6%,纯度97.5%。对比例1-2头孢曲松钠的合成与实施例1-1的合成工艺相同,区别仅在于:步骤1)中,alcl3-bf3碳酸二甲酯复合催化剂中alcl3和bf3的摩尔比为1:1。得到7-act313g,收率:82.5%,纯度97.9%。对比例1-3头孢曲松钠的合成与实施例1-1的合成工艺相同,区别仅在于:步骤3)中,结晶方式为:降温至5~8℃,滴加析晶溶剂丙酮300ml,滴加时间4h,滴加完毕后,控制转速在80~100转/分钟,析晶2h,析晶结束后抽滤,用丙酮30ml淋洗滤饼两次,真空干燥得到头孢曲松钠水合物185g,纯度98.1%,收率90.7%,d9010~120μm。对比例1-4头孢曲松钠的合成与实施例1-1的合成工艺相同,区别仅在于:整个反应过程不避光。实验例实验例1头孢曲松钠晶体的晶型鉴定1.1)xrd检测测试仪器:d8advance型x射线衍射仪(德国布鲁克公司)。试验条件:cu靶,kα辐射,管压40kv,发散狭缝1.0mm,反散射狭缝1.0mm,接收狭缝0.1mm,扫描速度4°/min,扫描范围2°~50°,步长0.02°,每步计时0.1s,样品制备:取样品置于样品架上直接压平后,于x射线衍射仪中测定,得到x射线衍射图谱。样品:实施例1-1、头孢曲松标准品(中国药品生物制品检定所,批号:130480-201504)。x射线衍射图谱如图1和图2,相应的特征标记峰值如下:如图1所示,实施例1-1头孢曲松钠样品特征峰为:11.72±0.2°,15.99±0.2°,18.72±0.2°,19.85±0.2°,22.75±0.2°,23.08±0.2°,24.79±0.2°,27.35±0.2°,28.12±0.2°,34.64±0.2°,35.44±0.2°,其中,“±0.2”为允许的测量误差范围。如图2所示,头孢曲松标准品(中国药品生物制品检定所,批号:130480-201504),特征峰为:(11.12±0.2°,12.45±0.2°,15.41±0.2°,18.30±0.2°,18.81±0.2°,19.93±0.2°,21.13±0.2°,22.61±0.2°,23.77±0.2°,24.53±0.2°,25.18±0.2°,26.79±0.2°,27.69±0.2°,28.24±0.2°,29.49±0.2°,30.72±0.2°,33.74±0.2°。1.2)样品dsc检测结果如图3头孢曲松钠的dsc曲线显示,不存在熔点峰,在269.8℃附近有一尖锐的分解放热峰,放热峰前的小峰是头孢曲松钠脱出结晶水吸热峰(头孢曲松钠的脱水温度为162.9℃)。实验例2头孢曲松钠晶体品质测定2.1)长期试验根据2010年版药典头孢曲松钠项下规定测定实施例1-1~实施例1-6、对比例1-1~对比例1-4制得的头孢曲松钠的含量杂质,储存条件为25±2℃,相对湿度60±10%,结果如下表1:表1注:“未检出”是指质量含量低于0.01%。2.2加速试验)取实施例样品,按照市售包装,在温度40±2℃,相对湿度75±5%的条件下放置6个月,分别在第1、2、3、6和12个月末取样检测,结果如下表2所示。表2从加速试验结果可以看出,本发明所制备的头孢曲松钠晶型各项指标的检测无显著变化,说明该晶型的稳定性较好,并且明显优于市售的罗氏产品,以及专利cn102875574制备的晶型(见发明专利cn102875574a中稳定性试验数据)。2.3)引湿性试验样品:实施例1-1、中检院(130480-201504)、罗氏(批号sh0299)、和中外制药株式会所(16l010a)头孢曲松钠晶体。测试方法:取干燥的具塞玻璃称量瓶(外径为50mm,高为15mm),于试验前一天置于人工气候箱(设定温度为25℃±1℃,相对湿度为80%±2%)内,精密称定重量(m1)。取供试品适量,平铺于上述称量瓶中,供试品厚度约为1mm,精密称定重量(m2)。将称量瓶敞口,并与瓶盖同置于上述恒温恒湿条件下24小时。盖好称量瓶盖,精密称定重量(m3)。增重百分率=(m3-m2)/(m2-m1)×100%。引湿性试验结果见表3:表3通过表3可以看出,本发明中方法制备得到的头孢曲松钠晶体具有较现有技术中晶体更低的引湿性,利于晶体的长期储存。实验例3抑菌性测试样品:实施例1-1、中检院(130480-201504)、罗氏(批号sh0299)和中外制药株式会所(厂家批次16l010a)头孢曲松钠晶体。测试方法:肉汤稀释法(培养基为mueller-hinton肉汤,ph7.2-7.4)。供试品制备及菌液接种:取无菌试管(13*100mm)13支,排成一排,除第1只加入1.6mmmh肉汤外,其余每管加入mh肉汤1ml,在第1管加入不同来源的头孢曲松钠(浓度均为320ug/ml)0.4ml混匀,然后吸取1ml至第2管,混匀后再吸取1ml至第3管,如此连续倍比稀释至第11管,并从第11管中吸取1ml弃去,第12管为不含药物的生长对照。此时各管药物浓度依次为64、32、16、8、4、2、1、0.5、0.25、0.125、0.0625ug/ml。然后在每管内加入金黄色葡萄球菌菌种(atcc29213)各1ml,使每管最终菌液浓度约为5*105cfu/ml。第1管至第11管药物浓度分别为32、16、8、4、2、1、0.5、0.25、0.125、0.0625、0.03125ug/ml。将接种好的稀释管塞好塞子,至35℃普通空气孵箱中孵育16-20小时。以肉眼观察,药物最低浓度管无细菌生长者,即为受试菌的mic。测试结果见表4:表4样品最低抑菌浓度(mic,ug/ml)实施例1-11-2标准品4-8罗氏2-4中外制药株式会所4-8实验例4药理研究实验例4.1样品:采用实施例2-1中制得的制剂。试验组:选用新西兰白兔20只(雌雄各半),采用静脉导管插入术在白兔右心室内接种肠球菌造模,得到肠球菌心内膜炎模型兔;以200mg/kg(以头孢曲松计)的体重尾静脉注射给药,每日1次,连续给药3天后,检测炎症情况(指标:c反应蛋白。无炎症:c反应蛋白:<15ng/ml;略有炎症:c反应蛋白:15ng/ml-35ng/ml;严重炎症:c反应蛋白:>35ng/ml)。对照组:选用健康新西兰白兔20只(雌雄各半),不做任何处理。结果如下:试验组:无炎症18只,略有炎症2只,严重炎症0只。对照组:无炎症20只,略有炎症0只,严重炎症0只。在试验过程中,新西兰白兔未表现出急躁、骚鼻、喷嚏等过敏性状。实验例4.2感染性心内膜炎模型的建立:选用健康昆明鼠100只(雌雄各半),采用静脉静脉导管插入术在小鼠左心室内接种金黄色葡萄球菌造模,得到模型小鼠。试验组:将实施例1-1中制备的粉针剂以500mg/kg的剂量尾静脉注射入模型建立1天后的模型试验组小鼠体内,每日注射1次,注射7天后脊椎脱臼法处死小鼠,对小鼠心脏进行观察。试验组包括30只雄鼠和30只雌鼠。对照组:向小鼠体内注射生理盐水,注射体积、注射频率同与试验组。对照组包括20只雄鼠和20只雌鼠。对小鼠的药理研究结果如下表5所示:表5实验例5临床试验将实施例2-1中制备的粉针剂进行临床试验,选定患有细菌性内膜炎患者100例,年龄25~70周岁之间。试验组(50例):静脉滴注注射用头孢曲松钠舒巴坦复方制剂1g(以头孢曲松钠计),30分钟滴完,检测血药浓度。连续静脉滴注7天,7天后检测炎症情况(指标:降钙素原pct。无炎症:pct:<0.5ng/ml;略有炎症:pct:0.5ng/ml-2ng/ml;出现严重炎症:pct:>2ng/ml);对照组(50例):不做任何处理。评价标准:显效:无炎症;有效:略有炎症;无效:出现严重炎症。实验结果为:经过抽血留样,并检测血样中头孢曲松钠和舒巴坦钠的血药浓度,计算受试者的平均值,绘制药时曲线,如图4和图5所示。可见,药时曲线上,头孢曲松钠的峰浓度(cmax)为150.0μg/ml,清除半衰期约为7.2h;舒巴坦钠峰浓度(cmax)为43.1μg/ml,清除半衰期约为66min。经统计,结果如下表6:表6组别显效有效无效试验组34133对照组0743其中,在治疗过程中,未发现过敏事件。以上结合具体实施方式和范例性实例对本发明进行了详细说明,不过这些说明并不能理解为对本发明的限制。本领域技术人员理解,在不偏离本发明精神和范围的情况下,可以对本发明技术方案及其实施方式进行多种等价替换、修饰或改进,这些均落入本发明的范围内。本发明的保护范围以所附权利要求为准。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1