橄榄状N-甲基-4-硝基邻苯二甲酰亚胺晶体及其制备方法与流程
本发明涉及一种n-甲基-4-硝基邻苯二甲酰亚胺晶体及其制备方法,属于精细化学品合成技术领域。
背景技术:
n-甲基-4-硝基邻苯二甲酰亚胺晶体是合成聚醚酰亚胺、聚酯酰亚胺、聚苯酰亚胺等聚酰亚胺材料的一类重要的中间体。其结构如下:
许多文献公开报道了该物质的制备方法(参见:郑凯,姚成.4-硝基-n-甲基-邻苯二甲酰亚胺合成新工艺[j].;石油化工,2004,33(2):145-148.郑凯,姚成.4-硝基-n-甲基-邻苯二甲酰亚胺的合成新工艺研究[j].江苏化工,2003,31(6):39-41.),其现有的技术如下:将n-甲基-4-硝基邻苯二甲酰亚胺溶解于浓硫酸中,向体系中滴加发烟硝酸,控制反应温度在10℃以下,维持几小时后,将体系升温至80℃以下反应几小时,可以获得80%以上的产品收率。
本申请的发明人在实际研究过程中发现,该方法存在着很大的问题,一是在硝化反应在低温下进行可能会出现积热现象,反而会带来安全隐患;二是产品本身收率偏低;三是该方法制备的n-甲基-4-硝基邻苯二甲酰亚胺固体堆积密度小,在合成下游产品时,出现加料困难、下游产品收率偏低等现象。
技术实现要素:
本发明的宗旨就是为了解决硝化反应安全问题和下游产品应用时加料困难、收率偏低的问题。
本发明采用的技术方案如下:
一种制备橄榄状n-甲基-4-硝基邻苯二甲酰亚胺晶体的方法,包括以下步骤:a)n-甲基邻苯二甲酰亚胺在浓硫酸体系中与发烟硝酸反应,制得n-甲基-4-硝基邻苯二甲酰亚胺的浓硫酸溶液;b)将n-甲基-4-硝基邻苯二甲酰亚胺的浓硫酸溶液倾倒在低温水中搅拌,析出n-甲基-4-硝基邻苯二甲酰亚胺固体,并用水将固体洗涤至滤液为中性并(优选在常压下)干燥;c)将干燥后的n-甲基4-硝基邻苯二甲酰亚胺固体投入到溶解磷酸酯和有机碱的酯类溶剂中进行回流并冷却,将混合物经过过滤和干燥(优选真空干燥),得到橄榄状的n-甲基-4-硝基邻苯二甲酰亚胺晶体。
进一步地,所述浓硫酸为浓度在93~98wt%;所述发烟硝酸浓度为85~98wt%,优选95~98wt%。
进一步地,n-甲基-4-硝基邻苯二甲酰亚胺与浓硫酸和发烟硝酸的质量比为1∶2~5∶0.2~3,优选1∶3~3.6∶0.3~1.5,更优选1∶3~3.1∶0.4~1.1。
进一步地,步骤a)中,将n-甲基-4-硝基邻苯二甲酰亚胺固体加入到开启搅拌的浓硫酸中,逐渐加热至溶解,将发烟硝酸逐渐滴加至反应体系中,反应一段时间后,降温至常温,所述加热溶解n-甲基邻苯二甲酰亚胺温度选自30~60℃,优选35~45℃;所述滴加硝酸时控制的体系温度为30~100℃;所述反应时间为1.5~5小时,优选2~3小时。
进一步地,步骤b)中,所述低温水选自冰水混合物以及0~15℃的水,优选冰水混合物;所述低温水的质量为n-甲基-4-硝基邻苯二甲酰亚胺理论产量的2.5~5倍,优选3~4倍;所述洗涤后滤液为中性的ph值为6~8;所述干燥温度为80~120℃,优选90~100℃。
进一步地,步骤c)中,所述回流洗涤的溶剂选自沸点≤154℃的酯类,包括但不限于乙酸乙酯、乙酸甲酯、乙酸异丙酯、乳酸乙酯中的一种或多种;所述磷酸酯类选自三聚磷酸酯、磷酸三丁酯、亚磷酸三苯酯、多聚磷酸等中的一种或多种;所述有机碱为有机杂环碱性化合物,优选选自吡啶、噻吩、吲哚、哌嗪、甲基吡啶等有机杂环物质。
进一步地,所述磷酸酯、有机碱和酯溶剂的质量比可以为1~5∶3~5∶90~96,优选2~5∶3~4∶91~95。
进一步地,回流洗涤过程中的固体加入量为总体系的10~35wt%,优选25~30wt%;回流时间为1~5小时,优选2~3小时;所述过滤方式为普通过滤、真空抽滤、压滤、离心等方式;所述真空干燥温度为70~140℃,优选75~120℃。所述真空干燥压力为-0.1~-0.095mpa。
本发明进一步涉及通过上述方法获得的橄榄状的n-甲基-4-硝基邻苯二甲酰亚胺晶体。
本发明的有益效果在于:
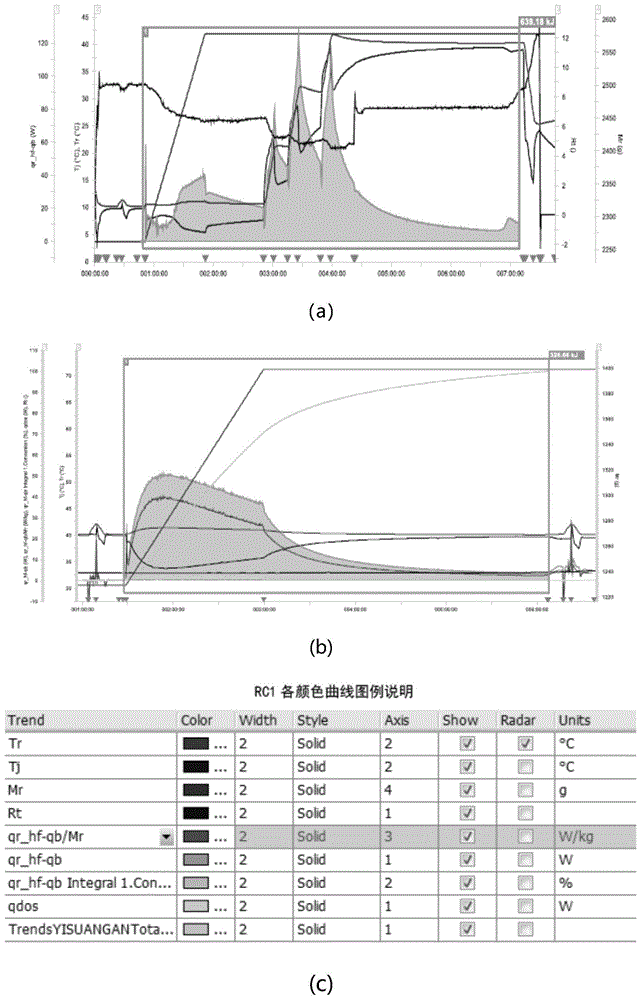
1、避免了低温硝化过程的热量积累,能够加快反应速率,提高反应过程的安全性,参见附图1。
2、提高了n-甲基-4-硝基邻苯二甲酰亚胺的产物收率。
3、使用磷酸酯和有机杂环碱性化合物为晶体成型助剂,有利于晶体成型,提高产物堆积密度,有利于下游产品合成,提高下游产品的收率,参见附图2和附图3。
附图说明
图1为对比例1和实施例1低温硝化和高温硝化的rc1量热测试图,其中(a)为对比例1的rc1量热测试图,(b)为实施例1的rc1量热测试图,(c)为rc1各颜色曲线图例说明。
图2为传统方法和本发明方法的晶体形状比较图,其中(a)为橄榄状的实施例1产物晶体形状,(b)为大小无规则形状的对比例1产物晶体形状。
图3为采用本方法制得的晶体形状的xrd谱图,其中(a)为橄榄状的实施例1产物晶体xrd谱图,(b)为大小无规则形状的对比例1产物晶体形状xrd谱图。
具体实施方式
以下结合实施例对本发明做进一步的说明,以下实施例只是作为对本发明的说明,不用于限制本发明的保护范围。
实施例1
将161gn-甲基邻苯二甲酰亚胺固体加入到开启搅拌的98wt%的300g浓硫酸中,将体系加热至40℃,直至n-甲基邻苯二甲酰亚胺完全溶解。称取98wt%的发烟硝酸65g,通过滴液漏斗将发烟硝酸逐渐加入体系,体系通冷却水降温移走热量,保持体系温度在40℃。当硝酸滴加完毕后,保持40℃,反应2.5小时。
将反应体系降至常温,将其倾倒在具有搅拌下的618g冰水混合物中析出。将产物n-甲基-4-硝基邻苯二甲酰亚胺固体过滤并用水洗涤至ph=7后,将产物在90℃下进行常压干燥。
将干燥后的产物加入洗涤提纯溶剂中,其溶剂组成为磷酸三丁酯∶吡啶∶乙酸乙酯=4∶4∶92,体系中n-甲基-4-硝基邻苯二甲酰亚胺含量占28%;在搅拌下加热回流,回流时间2.6小时;将体系降温至常温,将固体过滤,并用乙酸乙酯洗涤一遍后,在-0.098mpa下、80℃下干燥,即得到橄榄状n-甲基-4-硝基邻苯二甲酰亚胺晶体。收率为93%。堆积密度0.60~0.65g/ml。
实施例2
将161gn-甲基邻苯二甲酰亚胺固体加入到开启搅拌的93wt%的326g浓硫酸中,将体系加热至40℃,直至n-甲基邻苯二甲酰亚胺完全溶解。称取97wt%的发烟硝酸71.44g,通过滴液漏斗将发烟硝酸逐渐加入体系,体系通冷却水降温移走热量,保持体系温度在45℃。当硝酸滴加完毕后,保持60℃,反应2小时。
将反应体系降至常温,将其倾倒在具有搅拌下的515g冰水混合物中析出。将产物n-甲基-4-硝基邻苯二甲酰亚胺固体过滤并用水洗涤至ph=7后,将产物在100℃下进行常压干燥。
将干燥后的产物加入洗涤提纯溶剂中,其溶剂组成为三聚磷酸酯∶噻吩∶乙酸甲酯=1∶3∶96,体系中n-甲基-4-硝基邻苯二甲酰亚胺含量占25%;在搅拌下加热回流,回流时间1小时;将体系降温至常温,将固体过滤,并用乙酸甲酯洗涤一遍后,在-0.099mpa下、70℃下干燥,即得到橄榄状n-甲基-4-硝基邻苯二甲酰亚胺晶体。收率为91.5%。堆积密度0.60~0.65g/ml。
实施例3
将161gn-甲基邻苯二甲酰亚胺固体加入到开启搅拌的96wt%的306g浓硫酸中,将体系加热至30℃,直至n-甲基邻苯二甲酰亚胺完全溶解。称取95wt%的发烟硝酸66.3g,通过滴液漏斗将发烟硝酸逐渐加入体系,体系通冷却水降温移走热量,保持体系温度在30℃。当硝酸滴加完毕后,保持30℃,反应3小时。
将反应体系降至常温,将其倾倒在具有搅拌下的824g冰水混合物中析出。将产物n-甲基-4-硝基邻苯二甲酰亚胺固体过滤并用水洗涤至ph=7后,将产物在80℃下进行常压干燥。
将干燥后的产物加入洗涤提纯溶剂中,其溶剂组成为亚磷酸三苯酯∶吲哚∶乳酸乙酯=5∶5∶90,体系中n-甲基-4-硝基邻苯二甲酰亚胺含量占35%;在搅拌下加热回流,回流时间2小时;将体系降温至常温,将固体过滤,并用乳酸乙酯洗涤一遍后,在-0.095mpa下、140℃下干燥,即得到橄榄状n-甲基-4-硝基邻苯二甲酰亚胺晶体。收率为94%。堆积密度0.60~0.65g/ml。
实施例4
将161gn-甲基邻苯二甲酰亚胺固体加入到开启搅拌的98wt%的360g浓硫酸中,将体系加热至60℃,直至n-甲基邻苯二甲酰亚胺完全溶解。称取85wt%的发烟硝酸111g,通过滴液漏斗将发烟硝酸逐渐加入体系,体系通冷却水降温移走热量,保持体系温度在35℃。当硝酸滴加完毕后,保持30℃,反应5小时。
将反应体系降至常温,将其倾倒在具有搅拌下的1030g冰水混合物中析出。将产物n-甲基-4-硝基邻苯二甲酰亚胺固体过滤并用水洗涤至ph=7后,将产物在120℃下进行常压干燥。
将干燥后的产物加入洗涤提纯溶剂中,其溶剂组成为多聚磷酸∶甲基吡啶∶乙酸异丙酯=5∶4∶91,体系中n-甲基-4-硝基邻苯二甲酰亚胺含量占10%;在搅拌下加热回流,回流时间3小时;将体系降温至常温,将固体过滤,并用乙酸异丙酯洗涤一遍后,在-0.095mpa下、75℃下干燥,即得到橄榄状n-甲基-4-硝基邻苯二甲酰亚胺晶体。收率为84%。堆积密度0.60~0.65g/ml。
实施例5
将161gn-甲基邻苯二甲酰亚胺固体加入到开启搅拌的98wt%的300g浓硫酸中,将体系加热至35℃,直至n-甲基邻苯二甲酰亚胺完全溶解。称取98wt%的发烟硝酸65.6g,通过滴液漏斗将发烟硝酸逐渐加入体系,体系通冷却水降温移走热量,保持体系温度在100℃。当硝酸滴加完毕后,保持100℃,反应1.5小时。
将反应体系降至常温,将其倾倒在具有搅拌下的721g冰水混合物中析出。将产物n-甲基-4-硝基邻苯二甲酰亚胺固体过滤并用水洗涤至ph=7后,将产物在95℃下进行常压干燥。
将干燥后的产物加入洗涤提纯溶剂中,其溶剂组成为磷酸三丁酯∶哌嗪∶乙酸乙酯=2∶3∶95,体系中n-甲基-4-硝基邻苯二甲酰亚胺含量占30%;在搅拌下加热回流,回流时间5小时;将体系降温至常温,将固体过滤,并用乙酸乙酯洗涤一遍后,在-0.097mpa下、120℃下干燥,即得到橄榄状n-甲基-4-硝基邻苯二甲酰亚胺晶体。收率为94%。堆积密度0.60~0.65g/ml。
对比例1
将161gn-甲基邻苯二甲酰亚胺固体加入到开启搅拌的98wt%的300g浓硫酸中,将体系加热至40℃,直至n-甲基邻苯二甲酰亚胺完全溶解后,将体系温度降低至10℃以下。称取98wt%的发烟硝酸65g,通过滴液漏斗将发烟硝酸逐渐加入体系,体系通冷却水降温移走热量,保持体系温度在10℃。当硝酸滴加完毕后,保持10℃,反应3小时。逐渐采用阶梯升温方式升温至40℃,反应3小时。
将反应体系降至常温,将其倾倒在具有搅拌下的618g冰水混合物中析出。将产物n-甲基-4-硝基邻苯二甲酰亚胺固体过滤并用水洗涤至ph=7后,将产物在90℃下进行常压干燥,得到无规则粒径大小不一的n-甲基-4-硝基邻苯二甲酰亚胺晶体。收率为80%。堆积密度0.35~0.40g/ml。
应用实施例1
在1000ml的四口烧瓶中通入高纯氮气保护后,开启搅拌,以此加入8.0g氢氧化钠、22.8g双酚a和570g邻二甲苯,加热至回流温度144℃,加热反应4小时。之后逐渐将分水器中的水分出反应体系,待分水器中看不到水滴时,将反应体系中所有邻二甲苯蒸出体系,降温至100℃以下并始终保持氮气畅通。
称量的41.2g以实施例1方法制备的n-甲基-4-硝基邻苯二甲酰亚胺加热溶于n,n-二甲基乙酰胺中,在反应体系中快速地加入将溶液加入,并将反应体系升温至155℃,反应6小时后,降温至常温。
将反应后的溶液加入用乙醇中进行析出、过滤,用乙醇洗涤干净并用水洗涤乙醇和盐,将固体在100℃常压,得到白色的n-甲基-n′-甲基-3,3′,4,4′-双酚a二醚双邻苯二甲酰亚胺固体,摩尔收率94.2%。
应用对比例1
将对比例1方法制备的n-甲基-4-硝基邻苯二甲酰亚胺固体用于n-甲基-n′-甲基-3,3′,4,4′-双酚a二醚双邻苯二甲酰亚胺的合成,其合成方法与应用实施例1的方法相同。得到的n-甲基-n′-甲基-3,3′,4,4′-双酚a二醚双邻苯二甲酰亚胺产物收率为75%。
- 还没有人留言评论。精彩留言会获得点赞!