一种两面针碱衍生物的合成方法及其抗肿瘤应用与流程
本发明所属有机合成技术领域,特别涉及一种两面针碱衍生物的制备方法及其抗肿瘤应用。
背景技术:
两面针(zanthoxylumnitidum)又名入地金牛、蔓椒、双面针等,为芸香科花椒属藤本植物。在我国主要分布于华南地区。其中以广西的资源最为丰富,为两面针的道地药材产区。
两面针碱(nitidine)主要从两面针植物的根部提取分离得到。从结构上看它是由四个环组成,苯环和菲啶环并联在一起,形成共轭的平面结构。近年来的研究发现,两面针碱对肝癌、肺癌、乳腺癌、鼻咽癌等都有显著的功效,成为开发新药的重要先导化合物,吸引了许多化学家对两面针碱类化合物的研究。但由于两面针碱本身水溶性差、生物利用率低,大规模生成效率低、成本高,限制了两面针碱的成药性,不利于系统研究和临床使用。因此,对其化学结构进行改造成为了人们关注的焦点,特别是定点取代衍生物。本发明希望通过结构改造和修饰克服这些缺陷,改善成药性,提高抗肿瘤活性,降低毒性。两面针碱的b环上n-5、c-6位是重要的可修饰位,在保留两面针碱异喹啉环结构的基础上,简化其母核,设计合成一些结构简化的类似物,能够获得一系列具有抗肿瘤、抗炎、抗菌、抗hiv等活性的衍生物。
技术实现要素:
本发明的目的在于提供一种新型两面针碱衍生物合成方法及抗肿瘤应用。
本发明的技术方案是这样实现的:
一种两面针碱衍生物6-(2-二甲氨基乙基)氨基-3,8,9-三甲氧基菲啶(6),其结构式如下:
本发明提供了如下所述的两面针碱衍生物6-(2-二甲氨基乙基)氨基-3,8,9-三甲氧基菲啶(6)的合成方法,包括以下步骤:
1)以2-溴-4,5-二甲氧基苯甲酸为起始原料,与间氨基苯基醚在有机溶剂中反应得到2-溴-4,5-二甲氧基-n-(3-甲氧基苯基)苯甲酰胺(1);
2)2-溴-4,5-二甲氧基-n-(3-甲氧基苯基)苯甲酰胺在有机溶剂中引入氮原子的保护基得到中间体(2);
3)中间体(2)在有机溶剂中发生偶联反应成环得到关键中间体3,8,9-三甲氧基-5-(4-甲氧基苄基)菲啶-6-酮(3);
4)关键中间体3,8,9-三甲氧基-5-(4-甲氧基苄基)菲啶-6-酮(3)在酸性条件下脱保护得到产物3,8,9-三甲氧基菲啶-6(5h)-酮(4);
5)3,8,9-三甲氧基菲啶-6(5h)-酮(4)在氯化剂的作用下发生氯代反应得到产物6-氯-3,8,9-三甲氧基菲啶(5);
6)6-氯-3,8,9-三甲氧基菲啶(5)发生取代反应得到目标产物6-(2-二甲氨基乙基)氨基-3,8,9-三甲氧基菲啶(6),合成路线见下所示:
在上述合成路线中,有机溶剂可以根据反应的需求,从n,n-二甲基甲酰胺(缩写:dmf)、二氯甲烷中选取。反应温度可根据反应类型适当选取。反应时间可通过薄层色谱监控原料情况得出。
本发明所述的两面针碱衍生物的合成方法,具体包括以下步骤:
1)取2-溴-4,5-二甲氧基苯甲酸,加入氯化亚砜进行反应,反应物除去溶剂得中间体酰氯,将酰氯用二氯甲烷溶解后加入间氨基苯甲醚,经亲核取代反应得到酰胺(1);
2)取酰胺(1)溶解于n,n-二甲基甲酰胺中,加入氢化钠进行反应,再加入4-甲氧基苄氯,所得产物经硅胶柱层析,得中间体(2);
3)取中间体(2)溶于n,n-二甲基甲酰胺中,加入醋酸钯、三(邻甲苯基)膦、碳酸钾在惰性气体气氛保护条件下反应,所得产物经硅胶柱层析,得化合物(3);
4)取化合物(3),加入三氟乙酸反应,所得产物经硅胶柱层析,得化合物4。
5)取化合物(4),加入三氯氧磷反应,所得产物经冰水、浓氨水处理,过滤得化合物(5);
6)取化合物(5),加入n,n-二甲氨基乙二胺,反应物减压蒸馏除去溶剂,洗涤,得到目标产物(6)。
上述合成反应步骤1)中,2-溴-4,5-二甲氧基苯甲酸和氯化亚砜的反应温度优选65℃条件下进行,反应优选采用加热回流的方式进行,亲核取代反应温度优选0℃到室温条件下进行,反应是否完全可采用薄层层析跟踪监测,在上述条件下,反应完全大约需要3h。反应结束后经萃取、浓缩、柱分离得到酰胺(1)。该步骤中2-溴-4,5-二甲氧基苯甲酸和间氨基苯基醚摩尔比优选为1:1.1,为避免反应剧烈,分批加入酰氯。该步骤中所得物质上硅胶柱层析时,优选的是用由体积比1:5的石油醚和二氯甲烷组成的混合溶剂洗脱剂洗脱,即得到中间体酰胺(1)。
上述合成反应步骤2)中,中间体酰胺(1)、氢化钠和4-甲氧基苄氯的摩尔比优选为1:2:3;上述反应优选温度为室温,反应是否完全可采用薄层层析跟踪监测,在上述条件下,反应至完全大约需要8h。该步骤中所得物质上硅胶柱层析时,优选的是用二氯甲烷洗脱剂洗脱,即得到中间体(2)。
上述合成反应步骤3)中,中间体(2)、醋酸钯、三(邻甲苯基)膦和碳酸钾的摩尔比优选为1:0.1:0.2:4;上述反应优选是在100℃下进行,气氛条件优选为氮气,反应优选采用加热回流的方式进行。在上述条件下,反应至完全大约需要10h。该步骤中所得物质上硅胶柱层析时,优选的是用由体积比2:1的石油醚和乙酸乙酯组成的混合溶剂洗脱剂洗脱,即得到化合物(3)。
上述合成反应步骤4)中,反应优选是在75℃下进行。
上述合成反应步骤5)中,反应优选的温度为105℃(反应是否完全可采用薄层层析跟踪监测),反应时间优选为2h。
上述合成反应步骤6)中,化合物(5)和n,n-二甲氨基乙二胺的摩尔比优选为1:0.04;反应优选的温度为104~105℃,反应时间优选为5h,气氛条件优选为氮气,反应优选采用加热回流的方式进行。
活性测试证明,本发明设计并合成得到的两面针碱衍生物(6)具有很好的抗肿瘤作用。
因此,本发明提供了两面针碱衍生物(6)用于制备抗肿瘤药物的用途。经活性测试证明该衍生物具有良好的抗肿瘤效果,有望用于临床作为抗肿瘤药物。此外,本发明的制备方法反应条件温和,原料易得,成本低廉,收率高,易于操作实施。
附图说明
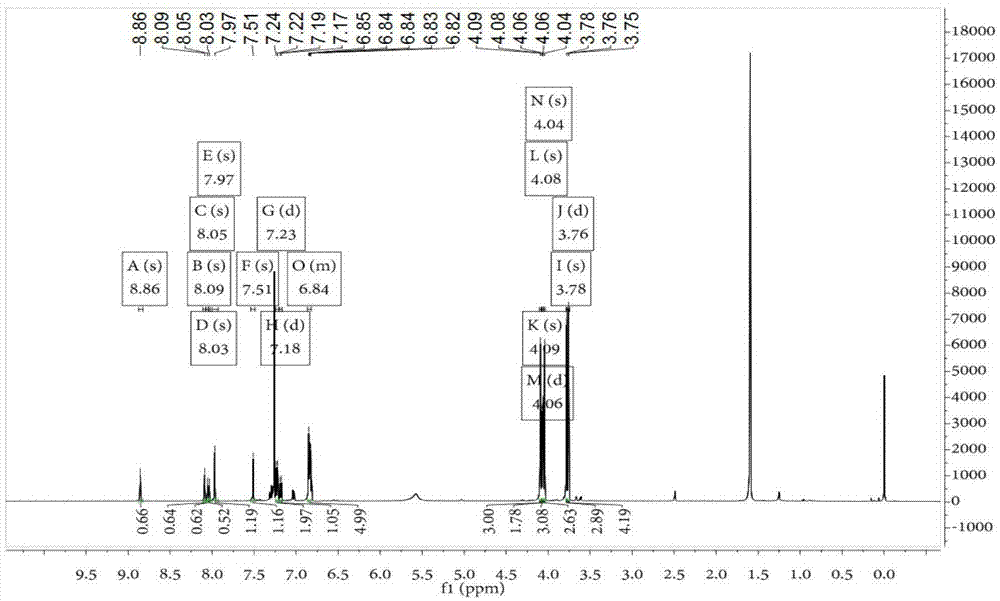
图1为本发明实施例1制备的3,8,9三甲氧基-5-(4-甲氧基苄基)菲啶-6-酮的1h-nmr图;
图2为本发明实施例1制备的3,8,9三甲氧基-5-(4-甲氧基苄基)菲啶-6-酮的13c-nmr图;
图3为本发明实施例1制备的3,8,9三甲氧基-5-(4-甲氧基苄基)菲啶-6-酮的esi-ms图;
图4为本发明实施例2制备的3,8,9-三甲氧基菲啶-6(5h)-酮的1h-nmr图;
图5为本发明实施例2制备的3,8,9-三甲氧基菲啶-6(5h)-酮的13c-nmr图;
图6为本发明实施例2制备的3,8,9-三甲氧基菲啶-6(5h)-酮的esi-ms图;
图7为本发明实施例3制备的6-氯-3,8,9-三甲氧基菲啶的1h-nmr图;
图8为本发明实施例3制备的6-氯-3,8,9-三甲氧基菲啶的13c-nmr图;
图9为本发明实施例3制备的6-氯-3,8,9-三甲氧基菲啶的esi-ms图;
图10为本发明实施例4制备的6-(2-二甲氨基乙基)氨基-3,8,9-三甲氧基菲啶的1h-nmr图;
图11为本发明实施例4制备的6-(2-二甲氨基乙基)氨基-3,8,9-三甲氧基菲啶的13c-nmr图;
图12为本发明实施例4制备的6-(2-二甲氨基乙基)氨基-3,8,9-三甲氧基菲啶的esi-ms图。
具体实施方式
下面结合具体实例和生物活性测试结果来进一步说明本发明,但不意味着限制本发明。
实施例1.3,8,9三甲氧基-5-(4-甲氧基苄基)菲啶-6-酮(3)的制备
1)中间体(1)的制备
在2-溴-4,5-二甲氧基苯甲酸(1.00g,3.83mmol)中加入10ml氯化亚砜,于65℃下加热回流1h。减压蒸馏除去溶剂,得白色固体酰氯;将间氨基苯甲醚(0.519g,4.21mmol)溶于2ml二氯甲烷中,0℃下向其加入5ml的n,n-二异丙基乙胺和溶解于二氯甲烷中的酰氯(在10min内分3次加入),反应1h后再转移至室温下搅拌1h。反应结束后,加入1m的盐酸淬灭,二氯甲烷萃取,有机相用饱和碳酸氢钠、饱和食盐水分别洗涤,经无水硫酸钠干燥后,减压浓缩,柱分离得到白色固体,即为2-溴-4,5-二甲氧基-n-(3-甲氧基苯基)苯甲酰胺(1),产率89.6%,比移值rf:0.42(展开剂为石油醚:二氯甲烷=1:5)。
2)中间体(2)的制备
将1mmol中间体(1)溶于10ml干燥的n,n-二甲基甲酰胺中,加入2eq的氢化钠,室温下搅拌30min,缓慢加入3eq的4-甲氧基苄氯,继续缓慢搅拌8h。反应结束后,加水淬灭,乙酸乙酯萃取,有机相合并后用水、饱和食盐水分别洗涤,无水硫酸钠干燥后减压浓缩,以二氯甲烷为洗脱剂进行柱分离,得中间体(2),产率为96%。
3)3,8,9-三甲氧基-5-(4-甲氧基苄基)菲啶-6-酮(3)的制备
将中间体(2)(1.47g,3.02mmol)溶解于15ml干燥的n,n-二甲基甲酰胺中,加入醋酸钯(0.068g,0.30mmol)、三(邻甲苯基)膦(0.185g,0.61mmol)和碳酸钾(1.671g,12.09mmol),氮气保护下,100℃加热回流10h。反应结束后冷却至室温,加水淬灭,二氯甲烷萃取,有机相水洗、饱和食盐水洗,无水硫酸钠干燥后,以石油醚和乙酸乙酯体积比2:1的洗脱剂进行柱分离,得白色固体,即为3,8,9-三甲氧基-5-(4-甲氧基苄基)菲啶-6-酮(3),产率:89.5%。m.p.176.8~178.9℃;ir(kbr,cm-1):3435,3131,1643,1607,1510,1466,1436,1400,1308,1241,1211,1177,1122,1029,825,775;1h-nmr(500mhz,cdcl3)δ8.86(s,1h),8.09(s,1h),8.05(s,1h),8.03(s,1h),7.97(s,1h),7.51(s,1h),7.23(d,j=8.7hz,2h),7.18(d,j=8.7hz,1h),6.86-6.82(m,5h),4.09(s,3h),4.08(s,2h),4.06(d,j=2.5hz,3h),4.04(s,3h),3.78(s,3h),3.76(d,j=2.8hz,4h);13c-nmr(126mhz,cdcl3)δ159.37,158.13,158.02,157.59,152.98,151.77,148.10,138.05,137.64,128.35,128.25,127.59,127.34,127.09,123.27,113.59,113.53,108.78,108.66,108.57,108.45,104.74,101.49,100.37,55.53,55.47,55.43,55.36,55.17,54.73,54.60,46.14,45.48;esi-msm/z:406.17([m+h]+)。
实施例2.3,8,9-三甲氧基菲啶-6(5h)-酮(4)的制备
取化合物(3)(0.70g,1.73mmol),加入2ml的三氟乙酸,75℃下加热回流10h。反应结束后冷却至室温,加乙酸乙酯、水淬灭,乙酸乙酯萃取,有机相水洗、饱和碳酸氢钠洗,无水硫酸钠干燥后,以二氯甲烷和乙酸乙酯体积比1:1的洗脱剂进行柱分离,得白色固体,即为3,8,9-三甲氧基菲啶-6(5h)-酮(4),产率:69.0%。m.p.267.4~269.1℃;ir(kbr,cm-1):3440,3160,1662,1612,1503,1404,1330,1258,1210,1176,1098,1044,877,840,802;1h-nmr(500mhz,cdcl3)δ7.99(d,j=8.9hz,1h),7.88(s,1h),7.51(s,1h),6.91-6.86(m,1h),6.74(s,1h),4.09(s,2h),4.05(d,j=9.4hz,3h),3.91(d,j=5.0hz,3h);13c-nmr(126mhz,cdcl3)δ160.56,159.42,153.18,148.40,137.30,129.26,124.42,117.70,113.38,111.11,109.69,107.64,103.40,99.09,55.86,55.31,55.07;esi-msm/z:286.12([m+h]+)。
实施例3.6-氯-3,8,9-三甲氧基菲啶(5)的制备
将0.02mmol3,8,9-三甲氧基菲啶-6(5h)-酮(4)置于反应瓶中,加入40ml的三氯氧磷,105℃下加热回流2h。反应结束后冷却至室温,小心倾入装有冰水的烧杯中,浓氨水逐滴加入至ph呈碱性,过滤沉淀,滤饼用水多次洗涤,以石油醚和二氯甲烷体积比1:1的洗脱剂进行柱分离,得白色固体,即为6-氯-3,8,9-三甲氧基菲啶(5),产率:82.67%。m.p.174.1~175.6℃;ir(kbr,cm-1):3443,3133,1617,1579,1503,1400,1313,1234,1213,1155,1113,1038,912,845;1h-nmr(500mhz,cdcl3)δ8.29(d,j=9.1hz,1h),7.77(s,1h),7.72(s,1h),7.47(d,j=2.6hz,1h),4.14(s,2h),4.09(s,2h),3.96(s,2h);13c-nmr(126mhz,cdcl3)δ160.75,154.46,150.99,150.39,145.16,131.52,123.64,119.62,119.16,118.66,109.59,107.69,102.44,57.04,56.97,56.38;esi-msm/z:304.08([m+h]+)。
实施例4.6-(2-二甲氨基乙基)氨基-3,8,9-三甲氧基菲啶(6)的制备
将5mmol的6-氯-3,8,9-三甲氧基菲啶(5)和0.2mmoln,n-二甲氨基乙二胺混合,在氮气保护下,104~106℃加热回流5h,反应结束后降至室温,旋蒸除去溶剂,固体加二氯甲烷溶解,有机相用5%氢氧化钠水溶液洗涤2次,再用水洗,无水硫酸钠干燥后得到固体粗产物,固体粗产物用二氯甲烷和甲醇体积比5:1的洗脱剂进行柱分离,得黄色固体,即为6-(2-二甲氨基乙基)氨基-3,8,9-三甲氧基菲啶(6),产率:59.0%。m.p.126.0~128.0℃;ir(kbr,cm-1):3414,3138,1617,1592,1400,1306,1209,1171,1039,841,801;1h-nmr(500mhz,cdcl3)δ8.08(d,j=8.8hz,1h),7.67(d,j=2.8hz,1h),7.34(s,1h),7.16(s,1h),6.94(dd,j=8.8,2.3hz,1h),4.06(d,j=11.9hz,7h),3.93(s,3h),3.84-3.78(m,2h),2.78-2.69(m,2h),2.37(s,6h);13c-nmr(126mhz,cdcl3)δ160.04,153.53,153.39,149.61,129.27,123.29,114.28,112.60,111.96,106.34,104.61,102.15,59.21,57.53,56.02,55.48,45.72,44.41,39.63;esi-msm/z:356.19([m+h]+)。
实施例5.两面针碱衍生物的抗肿瘤活性测试
将实施例1~实施例4所制备的两面针碱衍生物进行体外抗肿瘤活性测定。
选用细胞株:肝癌细胞hepg2、肺癌细胞a549和h460、鼻咽癌细胞cne1。
实验方法:将衍生物用二甲基亚砜溶解稀释成所需溶液的浓度。取对数生长期细胞,用胰酶消化,制成4×104~5×104个·ml-1细胞悬液,均匀接种于96孔培养板中,每孔100μl,为防止边缘效应,在96孔板四周每孔各加100μlpbs,置于co2培养箱中培养24h后,加入受试液,每孔10μl,培养48h。将3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐(mtt)加入96孔板中,每孔10μl,培养箱中反应4h。弃去上清液,加入二甲基亚砜,每孔100μl,微量振荡器上振荡5min,用酶标仪在570nm处测定每孔的吸光度,计算细胞的抑制率。
细胞抑制率(inhibitoryrate,100%)=(药物组od-空白组od)/(对照组od-空白组od)×100%
实验结果如下表所示。
实验数据表明,本发明两面针碱衍生物对肿瘤细胞增殖具有较强的作用,特别是6-(2-二甲氨基乙基)氨基-3,8,9-三甲氧基菲啶(6)具有高效的抗肿瘤活性。因此,可将其用于制备抗癌的候选药物。
- 还没有人留言评论。精彩留言会获得点赞!