阪崎克罗诺杆菌干粉化双重PCR检测试剂盒的制作方法
本发明涉及微生物分子检测试剂,具体涉及一种干粉化的阪崎克罗诺杆菌双重pcr检测试剂盒。
背景技术:
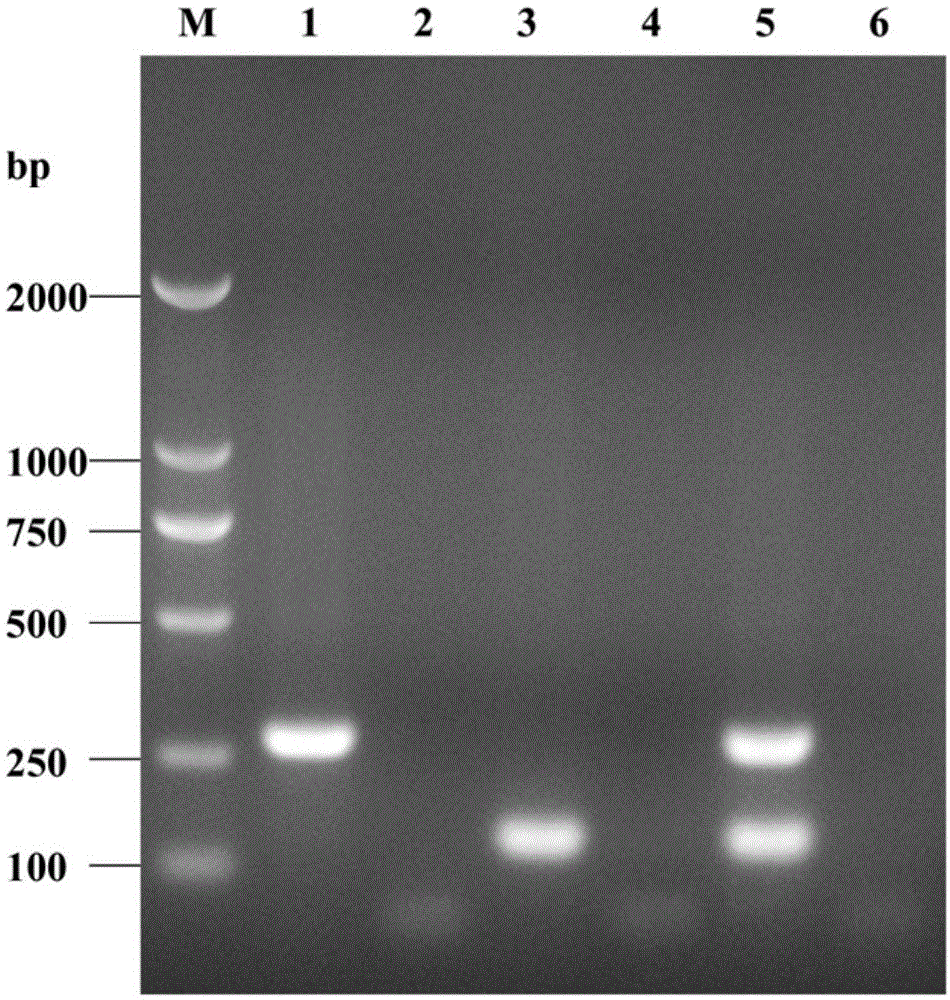
:阪崎克罗诺杆菌(cronobactersakazakii)是一种革兰氏阴性无芽孢杆菌,其具有周身鞭毛、动力以及产黄色素等特征,属于克罗诺杆菌属(cronobacter)。阪崎克罗诺杆菌是常见的食源性条件致病菌,已被国际食品微生物标准化委员会(internationalcommissiononmicrobiologicalspecificationsforfoods,icmsf)列为“严重危害特定人群,危害生命或慢性实质性后遗症或长期影响”的一种致病菌,其广泛分布于机体以及自然环境中,主要感染婴幼儿并引发坏死性小肠结肠炎、脑膜炎以及败血症及等严重病症,尤其是对免疫力低下、体重偏低、早产等婴幼儿危害更大,表现出极高的后遗症并发率和死亡率。另外,也有研究显示,该菌还会感染老年人或免疫力低下的成年人群。目前,对阪崎克罗诺杆菌具体致病机制以及污染源等尚不清楚,因此,加强对食品尤其是婴幼儿配方食品、乳和乳制品及其原料中阪崎克罗诺杆菌的监测与控制至关重要。当前,对食品中阪崎克罗诺杆菌的检测检验主要依据美国食药局(fda)推荐的相关方法,包括传统检测技术(如微生物分离培养鉴定、生化鉴定和血清学鉴定)、免疫学检测技术以及实时荧光定量pcr法等。目前国内主要以微生物分离培养和形态学鉴定为主、结合生化鉴定和血清学分型鉴定等通行方法进行食品微生物学相关检验,其初步鉴定一般需要2~3天,而最终完成鉴定报告则需5~7天。这些方法表现出检测周期长、操作复杂、仪器昂贵、成本高等不足,并且多局限在实验室检测,难以满足在基层等地区的检测需求。pcr技术发展至今已十分成熟,应用广泛,普通的pcr扩增仪、所需如taqdna聚合酶等试剂也在一般实验室中也能得到普及使用,在如gb4789.6-2016食品微生物学检验:致泻大肠埃希氏菌检验和gb4789.12-2016食品微生物学检验:肉毒梭菌及肉毒毒素检验等标准中均也提到可经pcr技术替代相关检测流程,同时针对阪崎克罗诺杆菌的pcr检测方法国内外研究很多,众多有关阪崎克罗诺杆菌的保守性基因也先后被报道,如ompa、cgca、its、16srdna和mms等,这些均为食源性致病微生物的pcr检测奠定了基础。另外,在进行pcr检测时,检测前一般都要配制相应的反应体系溶液,即一般为液体状,其成分较复杂,容易导致由于多次操作引起的误差,且易造成实验区间的交叉污染,加之其中部分主要试剂如taqdna聚合酶、dntps等需要冷冻储存,而高温长途运输和使用时反复冻融,均会影响其反应效率,同时也增加了储存、运输成本。另外,在相关报道中,一般采用普通单重pcr检测阪崎克罗诺杆菌,其普遍存在特异性差、产物目标条带无法通过根据电泳亮带有效辨别(可能出现非特异性条带与目标条带大小相当)等弊端,而采用有效的多重pcr检测则可大大提高其特异性,并且其产物目标条带更便于观察、识别。但多重pcr相对于单重pcr,其引物设计、筛选的难度也会大大增加,诸如:①用于引物设计的具体dna片段的选取需有效可行;②不同对引物的退火温度需要相近;③扩增片段大小相差需控制在100~200bp为宜;④多条引物之间需避免非特异性结合;⑤一对引物中的一条不能与其他对的引物一同起作用而产生非特异性扩增。同时多重pcr反应体系也较单重pcr反应体系更为复杂,在其反应体系优化测试中,需逐个测试并兼顾不同反应成分的浓度(如dntps、taqdna聚合酶以及引物浓度等),以确保获取更高的扩增效率,期间如遇到多重引物扩增效率不佳,则需重新设计整套多重引物,并再次进行其反应体系优化测试。目前市场上阪崎克罗诺杆菌检测相关的pcr检测试剂盒较少,另外在其pcr检测具体流程、样品前处理方式、检测基因特异性以及检测结果符合率不高等方面尚有不足。技术实现要素:本发明的目的在于克服现有技术的不足,提供一种阪崎克罗诺杆菌干粉化双重pcr检测试剂盒。本发明所采取的技术方案是:用于阪崎克罗诺杆菌检测的双重pcr引物组,其序列如下:引物对1:f1:gctaggccccggctagcatgcc和r1:cgctagctagcgtcgcattgtcc引物对2:f2:gctagttcgctcgagcttggcc和r2:tgcgatcgtgctcgtgcccgcg。一种检测阪崎克罗诺杆菌的双重pcr试剂盒,其双重pcr引物组的序列如下:引物对1:f1:gctaggccccggctagcatgcc和r1:cgctagctagcgtcgcattgtcc引物对2:f2:gctagttcgctcgagcttggcc和r2:tgcgatcgtgctcgtgcccgcg。作为上述双重pcr试剂盒的进一步改进,双重pcr试剂盒包括干粉化检测试剂和复溶液,其中:粉化检测试剂的组成为:dna聚合酶、dntps、引物和冻干保护剂。作为上述双重pcr试剂盒的进一步改进,复溶液的组成为:4~16mmol/l(nh4)2so4、1~15mmol/lmgso4、10~25mmol/ltris-hcl、5~18mmol/lkcl以及0.5~0.8%tritonx-100。作为上述双重pcr试剂盒的进一步改进,还包括裂解液,裂解液含有:5~30mmol/ltris-hclph8.5、0.5~1.5mmol/ledta以及0.5~1.5%sds。作为上述双重pcr试剂盒的进一步改进,冻干保护剂的组成为:3~6质量份的海藻糖、0.01~0.1质量份的甘氨酸和0.1~1质量份的牛血清白蛋白。作为上述双重pcr试剂盒的进一步改进,引物对1和引物对2的摩尔用量比为(0.3~1.5):(0.1~1.2)。作为上述双重pcr试剂盒的进一步改进,干粉化检测试剂冻干前的组成为:0.1~0.6u/μltaqdna聚合酶、0.2~1.8mmol/ldntps、0.3~1.5μmol/l引物16srdna-f/16srdna-r、0.1~1.2μmol/l引物ompa-f/ompa-r以及冻干复合保护剂含3%~6%海藻糖(w/v)、0.01%~0.1%甘氨酸(w/v)、0.1%~1%牛血清白蛋白,余量为无菌超纯水。作为上述双重pcr试剂盒的进一步改进,还包括阳性对照和阴性对照,其中,阳性对照干粉为提纯的阪崎克罗诺杆菌基因组dna冻干粉,阴性对照干粉为提纯的非阪崎克罗诺杆菌基因组dna冻干粉。作为上述双重pcr试剂盒的进一步改进,干粉化检测试剂的制备方法包括使用液氮预冻,之后冷冻干燥。本发明的有益效果是:本发明的双重pcr引物,具有很好的特异性和扩增效率。本发明的检测试剂盒,具有很好的特异性和扩增效率,从收集dna样本到检测完成仅需3~4小时,经核酸电泳观察结果,简便,高效快速。本发明的检测试剂盒,通过对冻干保护剂进行优化,大大增加试剂稳定性,试剂的保质期时间可以延长到2年,且便于常温运输,节约运输成本,另外在使用时加入预先配置的液态复溶液即溶即用,减少溶液配置步骤,更加便捷、准确,并能有效减少试剂损耗以及气溶胶污染等,更利于长途运输。附图说明图1是本发明双重pcr引物扩增后的电泳结果;图2是部分标准菌株的特异性实验电泳图;图3是本发明双重pcr引物的纯基因组dna灵敏度实验电泳图;图4是本发明双重pcr引物的纯培养菌灵敏度实验电泳图。具体实施方式下面结合实施例,进一步说明本发明的技术方案。dna样品制备1.标准菌株和分离株dna样品制备:挑取各种标准菌株和分离株的新鲜培养菌落半环于30μl裂解液,充分悬浮后经99℃热裂解15min,12000r/min离心10min后取上清,即为粗提dna样品,-40℃保存备用;2.食品dna样品制备:根据gb4789.40-2016,从克罗诺杆菌属定性检验操作步骤5.2.2开始,挑取可疑菌落,悬浮到含30μl裂解液的无菌离心管中,充分悬浮菌体,轻弹管壁消除气泡,经99℃热裂解10min,12000r/min离心10min后取上清,即为粗提dna样品,-40℃保存备用。双重pcr检测引物组设计和有效性试验1.通过筛选获取克罗诺杆菌特异性靶基因,采用primerpremier6.0软件分析设计并经过自己技术优化得到本发明的引物对。引物序列信息见表1。2.以标准菌株atcc29544dna样品为阳性模板,以非阪崎克罗诺杆菌dna样品为阴性对照,分别进行16srdna和ompa基因片段的单重pcr扩增和双重pcr扩增,其中单重pcr扩增反应体系见表2,双重pcr扩增反应体系见表3,pcr反应程序见表4。表1、引物序列表表2、单重pcr扩增反应体系组成表组分加入体积(μl)10×pcrbuffer2dntps(10mmol/l)1.2taqdna聚合酶(5u/μl)1.0引物-f(10μmol/l)0.8引物-r(10μmol/l)0.8dna样品3.0无菌超纯水11.2合计20表3、双重pcr扩增反应体系组成表表4、pcr反应程序3.电泳结果观察:以上反应后每管中加入4μl6×loadingbuffer,混匀,后取8μl样品进行1.5%琼脂糖凝胶电泳(110v,30min),并在紫外仪下观察结果。结果如图1所示,图中:m表示markerdl2000ladder;条带1表示16srdna基因片段单独扩增,在283bp处出现明显的特异性条带;条带3表示ompa基因片段单独扩增,在142bp处出现明显的特异性条带;条带5表示16srdna和ompa基因片段双重pcr扩增,分别在142bp和283bp处出现明显的特异性条带;条带2,4,6分别表示以上各自对应的阴性对照,均无目的条带出现。综上,本发明中采用的2对检测引物、反应体系可同时有效检出阪崎克罗诺杆菌的16srdna和ompa基因。试剂盒的制备1.引物合成:按照表1中提供的本发明引物组序列合成引物,并定量配制。2.制备试剂冻干粉:按照表5所示在无菌环境下混合需要冻干处理的相关组分混合液,封装于容器中,置于液氮中进行快速冷冻,并按照常规冷冻干燥程序进行干燥,即可得到干粉化的检测试剂。表5、试剂盒干粉化检测试剂的组成表3.制备复溶液:按照表6所示在无菌环境下混合复溶液混合液,置于容器中。表6、复溶液组成表组分终浓度(nh4)2so410mmol/lkcl12mmol/lmgso412mmol/ltris-hcl(ph8.8)20mmol/ltritonx-1000.6%4.制备阳性对照干粉:向提纯的阪崎克罗诺杆菌基因组dna溶液分别加入3%的海藻糖(w/v),分装于容器中,同样经上述冻干程序制备阳性对照干粉。5.制备阴性对照干粉:向提纯的非阪崎克罗诺杆菌基因组dna溶液分别加入3%的海藻糖(w/v),分装于容器中,同样经上述冻干程序制备阴性对照干粉。6.6×loadingbuffer:含有30~50mmol/ledta、30%~45%甘油(v/v)、0.02%~0.12%二甲苯腈蓝ff(w/v)以及0.05%~0.1%溴酚蓝(w/v),置于容器中。7.配制裂解液:含有5~30mmol/ltris-hclph8.5、0.5~1.5mmol/ledta以及0.5~1.5%sds,置于容器中。8.将上述6种容器组装成试剂盒,提供使用说明书,封装。特异性实验:本发明双重pcr实验:选取阪崎克罗诺杆菌标准菌株、分离菌株和非阪崎克罗诺杆菌标准菌株、分离菌株,并根据实施例1制备其各自对应的dna样品,利用本试剂盒进行测试,经核酸电泳观察是否出现目标条带,并计算该检测符合率,其中,检测符合率=(本检测阳性结果即真实为阪崎克罗诺杆菌菌株数+本检测阴性结果即真实为非阪崎克罗诺杆菌菌株数)/被检测菌株数×100%。其中,选取的34株标准菌株信息见表7,538株分离菌株信息见表8。表7、标准菌株信息表编号名称编号名称atcc29544阪崎克罗诺杆菌nctc12900大肠埃希氏菌o157:h7atcc51329阪崎克罗诺杆菌cmcc(b)26003金黄色葡萄球菌cmcc(b)44102大肠埃希氏菌cmcc(b)51105痢疾志贺氏菌atcc25922大肠埃希氏菌cmcc(b)63303蜡样芽孢杆菌atcc14028鼠伤寒沙门氏菌cmcc(b)52204小肠结肠炎耶尔森氏菌cmcc(b)50094甲副伤寒沙门氏菌cmcc(b)54007单核细胞增生李斯特菌cmcc(b)50071伤寒沙门氏菌atcc33291空肠弯曲杆菌cmcc(b)51592宋内氏志贺氏菌atcc17802副溶血性弧菌cmcc(b)51572福氏志贺氏菌atcc13048产气肠杆菌atcc6633枯草芽孢杆菌cmcc(b)31001肺炎链球菌atcc25923金黄色葡萄球菌atcc33787溶藻弧菌atcc9027铜绿假单胞菌cmcc(b)24009奇异变形杆菌atcc29212粪链球菌atcc10231白色念珠菌atcc43864弗氏柠檬酸杆菌cmcc(b)26069表皮葡萄球菌atcc13124产气荚膜梭菌cmcc(b)46117肺炎克雷伯氏菌atcc8739大肠埃希氏菌atcc9763酿酒酵母atcc27562创伤弧菌atcc33152嗜肺军团菌表8、538株分离菌株信息表本发明检测结果显示,所有阪崎克罗诺杆菌标准菌株、分离菌株检测均在目标位置出现两条明显的特异性条带,而所有非阪崎克罗诺杆菌标准菌株、分离菌株检测均未在目标位置出现两条明显的特异性条带,该检测符合率为100%。其中部分的标准菌株检测电泳图见图2,图2中,m表示markerdl2000ladder;1~14分别表示atcc29544,atcc51329,cmcc(b)44102,atcc25922,atcc14028,cmcc(b)50094,cmcc(b)50071,cmcc(b)51592,cmcc(b)51572,atcc6633,atcc25923,atcc9027,atcc29212以及atcc43864。单重pcr对比例1:取本发明中获取的16srdna、ompa基因单重pcr扩增引物(见表1),对上述表7中34株标准菌株制备对应的dna样本,分别进行16srdna和ompa基因片段的单重pcr扩增,扩增反应体系和扩增条件见表2、4,扩增产物经琼脂糖凝胶电泳观察,并分别记录各自目的片段出现情况,即16srdna基因片段单重pcr扩增中,是否在283bp处出现明显条带;ompa基因片段单重pcr扩增中,是否在142bp处出现明显条带,计算相应的检测符合率。检测结果见表9,显示有部分非阪崎克罗诺杆菌标准菌株对应的样品均会出现16srdna和ompa基因片段目的条带,即为明显的非特异性扩增,其中,“+”表示出现明显目标条带;“-”表示均未出现目的条带,并分别计算16srdna和ompa基因片段单重pcr扩增符合率分别为47.1%和67.7%。表9、单重pcr检测结果单重pcr对比例2:取之前相关报道中给出的阪崎克罗诺杆菌特异性基因16srdna、ompa基因单重pcr扩增引物,分别记作16srdna-ff、16srdna-fr、ompa-ff和ompa-fr(见表10),对上述表7中34株标准菌株制备对应的dna样本,分别进行该单重pcr扩增,扩增反应体系和扩增条件见表11、12,扩增产物经琼脂糖凝胶电泳观察,并分别记录各自目的片段出现情况,即16srdna基因片段单重pcr扩增中,是否在149bp处出现明显条带;ompa基因片段单重pcr扩增中,是否在469bp处出现明显条带,同样如上述计算相应的检测符合率。检测结果见表13,显示有部分非阪崎克罗诺杆菌标准菌株对应的样品均会出现16srdna和ompa基因片段目的条带,即为明显的非特异性扩增,其中,“+”表示出现明显目标条带;“-”表示均未出现目的条带,并分别计算16srdna和ompa基因片段单重pcr扩增符合率分别为55.9%和52.9%。表10、对比pcr引物序列表表11、对比例单重扩pcr增反应体系组成表组分加入体积(μl)10×pcrbuffer2dntps(10mmol/l)1.0taqdna聚合酶(5u/μl)1.0引物-f(10μmol/l)0.6引物-r(10μmol/l)0.6dna样品3.0无菌超纯水11.8合计20表12、pcr反应程序表13、对比例单重pcr检测结果综上可见,相比较普通的单重pcr扩增检测,本发明的双重pcr扩增具有极好的特异性,能有效检出阪崎克罗诺杆菌。灵敏度试验1.纯dna测试:取阪崎克罗诺杆菌atcc29544新鲜培养物接种到100mltsb中,36℃±1℃培养24h±2h,后经细菌基因组dna提取试剂盒提取其基因组dna,并测定其浓度为2.1×102ng/μl。以上纯dna经梯度稀释至2.1×10-6ng/μl,后分别取各浓度的dna稀释液2μl经本试剂盒检测,经核酸电泳观察是否出现目标条带。2.纯培养菌测试:取阪崎克罗诺杆菌atcc29544新鲜培养物,经无菌生理盐水进行梯度稀释(稀释倍数分别为100、101、102、103、104、105、106、107),并分别取各个稀释菌液100μl螺旋接种到tsa平板进行培养和计数;分别取以上稀释菌液100μl,6000r/min离心5min,弃上清,加入10μl裂解液充分悬浮菌体沉淀,99℃加热裂解菌体10min,菌体裂解液12000r/min离心15min,取上清即为粗提的dna样品,后分别取2μl经本试剂盒检测,经核酸电泳观察是否出现目标条带,并通过平板菌落计数确定最低检验限。3.结果:如图3所示,m表示markerdl2000ladder;1~8分别表示dna稀释液为2.1×101ng/μl、2.1×100ng/μl、2.1×10﹣1ng/μl、2.1×10﹣2ng/μl、2.1×10﹣3ng/μl、2.1×10﹣4ng/μl、2.1×10﹣5ng/μl以及2.1×10﹣6ng/μl,在浓度为2.1×10﹣3ng/μl时,仍能扩增出清晰的两条目的条带,可见本发明的纯基因组dna检测灵敏度可达2.1×10﹣3ng/μl;如图4所示,m表示markerdl2000ladder;1~8分别表示纯培养菌稀释倍数分别为107、106、105、104、103、102、101、100,并参照gb4789.2-2016《食品微生物学检验菌落总数测定》,测定其在105稀释倍数处对应的菌落数为25cfu,而由图4可知在103稀释倍数处,仍能扩增出清晰的两条目的条带,可见本发明的纯培养菌检验限为2.5×103cfu/ml,而在以往相关报道中,pcr检测其最低检验限一般为100-101pg/μl(即10﹣3-10﹣2ng/μl)或是102-103cfu/ml级别。综上可见,本发明可以达到与以往报道相当的检测灵敏度,能有效检出阪崎克罗诺杆菌。稳定性试验保质期试验取标准菌株atcc29544新鲜培养物,经无菌生理盐水悬浮、稀释、计数,获取浓度约为2.5×103cfu/ml的稀释液,并取1ml以上稀释液,6000r/min离心10min后弃上清,加入30μl裂解液,充分悬浮后经99℃热裂解15min,12000r/min离心10min后取上清,即为该稀释液制备的阳性dna样品。根据实施例3中试剂冻干粉制备获取9例不同组分的试剂冻干粉,相应地组装成9种不同的试剂盒,存放于2~8℃环境中,每隔3个月,经以上制备的dna样品测试该9种试剂盒产品性能,以上每种试剂盒检测做10个平行试验,并计算其各自检测效率,其中,检测效率=阳性结果数/总检测数×100%。结果,见表14,采用例9复合冻干保护剂进行反应试剂的冻干,经过液氮预冻和冷冻干燥后,试剂冻干粉能保持较高的稳定性,最长的保质期时间为21~24个月,远高于同类型产品的12个月保质期。另外,除含5%海藻糖的复合保护剂有吸潮现象外,其余复合保护剂冻干成粉状制剂后在各自保质期范围内均保持良好形态,没有塌陷和镂空现象。表14、冻干保护剂的稳定性实验结果重复性试验:采用经上述例9给出的复合冻干保护剂配方制备的试剂冻干粉,并组装成相应的检测试剂盒(包括5个生产批次,分别为:20180412,20180509,20180615,20180711,20180810),取以上保质期试验中制备的阳性dna样品和本试剂盒中阴性对照样品分别进行测试,具体包括:①选取不同的操作地点、人员以及仪器(在单一操作条件下,其他操作条件均一致),测试同一批次检测试剂盒的稳定性;②在相同的操作环境,测试以上5个生产批次检测试剂盒的稳定性。以上每份反应均做10个平行测试,且以上各种检测重复率=检测符合数/检测总数×100%。结果见表15,在不同操作环境中,本试剂盒的检测重复率均为100%;见表16,不同生产批次的本试剂盒其检测重复率均为100%。表15、不同操作条件下的重复性实验结果表16、不同批次的重复性实验结果模拟运输试验采用上述重复性试验中生产的检测试剂盒5盒,置于预先放有冰袋的泡沫箱中,并将该泡沫箱置于42℃环境中储存总计120h,期间分别在每24h后取出1盒本试剂盒,经以上保质期试验中制备的阳性dna样品和本试剂盒中阴性对照样品分别进行测试。同样,以上每份反应均做10个平行测试,并计算各种检测符合率=检测符合数/检测总数×100%。结果,见表17,在120h内的模拟运输后,本试剂盒的检测符合率均为100%。表17、模拟运输试验结果常规食品中阪崎克罗诺杆菌的检测1.样品前处理:参照上述dna样品制备的操作制备食品dna样品。2.加样、反应:取出干粉化检测反应管,每管分别加入复溶液18μl,充分溶解,分别加入各食品dna样品2μl,其中,取1管作为阴性对照,1管作为阳性对照,分别加入对应的对照试剂2μl,充分混匀,经biometraprofessionalpcr仪进行扩增反应,程序为:94℃预变性5min;94℃变性30sec,57℃退火30sec,72℃延伸30sec,35个循环;72℃充分延伸5min。3.结果判读:反应后每管中加入4μl6×loadingbuffer,混匀,取8μl样品进行1.5%琼脂糖凝胶电泳,恒压110v,时间30min,并在紫外仪下观察结果,阴性对照孔无条带出现,阳性对照孔分别在142bp和283bp处出现特异性条带,待测样品反应孔若与阳性对照孔条带一致,则表示有阪崎克罗诺杆菌检出;反之,则无检出。sequencelisting<110>广东环凯微生物科技有限公司广东环凯生物科技有限公司<120>阪崎克罗诺杆菌干粉化双重pcr检测试剂盒<130>d-pcr<160>8<170>patentinversion3.5<210>1<211>22<212>dna<213>人工引物<400>1gctaggccccggctagcatgcc22<210>2<211>23<212>dna<213>人工引物<400>2cgctagctagcgtcgcattgtcc23<210>3<211>22<212>dna<213>人工引物<400>3gctagttcgctcgagcttggcc22<210>4<211>22<212>dna<213>人工引物<400>4tgcgatcgtgctcgtgcccgcg22<210>5<211>20<212>dna<213>人工引物<400>5caagtcgaacggtaacaggg20<210>6<211>18<212>dna<213>人工引物<400>6gtcccccactttggtccg18<210>7<211>22<212>dna<213>人工引物<400>7ggatttaaccgtgaacttttcc22<210>8<211>19<212>dna<213>人工引物<400>8cgccagcgatgttagaaga19当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1