放射性药物的制作方法
本发明涉及新型化合物、含有该化合物的放射性药物、以及该放射性药物的制备用药物等。
背景技术:
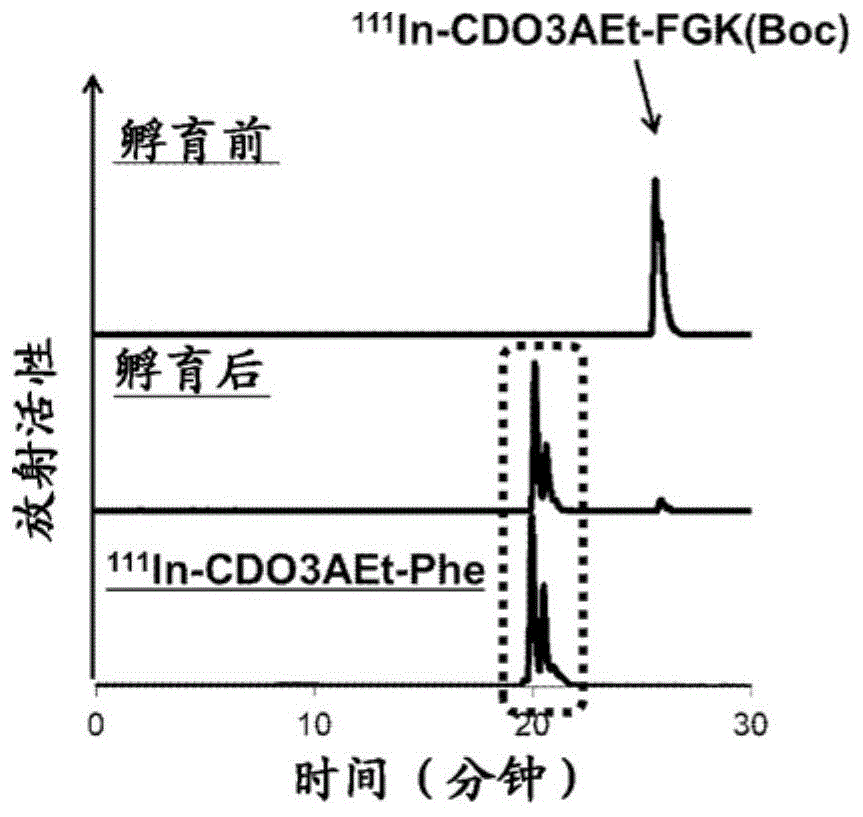
:放射性同位元素(ri)标记抗体等放射性药物能够利用抗体的高特异性和高亲和性而使ri选择性地积聚于肿瘤中。因此,应用于同位素治疗等放射线治疗、成像诊断中(非专利文献1)。然而,当将放射性药物施予至生物体时,除了向靶组织中的特异性积聚之外,还观察到向肾脏中的非特异性积聚。向肾脏中的放射活性积聚(以下也称为“肾积聚”)起因于:ri标记的小分子肽被肾脏摄入后、转运到溶酶体、并代谢后生成的放射性代谢物残存在肾脏中。与此相对,专利文献1中,作为能够自施予早期起减少向肾脏的积聚的放射性标记药物,报道了具有与nota(1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酸)等螯合剂结合的多肽部位的化合物及使用其的放射性药物。现有技术文献专利文献专利文献1:国际公开wo2017/150549号非专利文献非专利文献1:molecularoncology8:799-812,2014技术实现要素:发明所要解决的课题利用专利文献1的放射性药物,可以使用镓-67、锝-99m的放射性同位元素。然而,目前,尚未开发出能够应用于作为治疗用的放射性同位元素而通用的镥-177、钇-90、或应用于包括作为其伴随药物的铟-111等具有较大原子半径的原子在内的多种原子的标记药物。因此,本发明涉及能够获得放射性药物的化合物等,所述放射性药物能够利用包括具有较大原子半径的原子在内的多种原子进行标记,能够自施予早期起减少向肾脏的积聚。用于解决课题的手段本发明涉及以下的实施方式。〔1〕下述式(1)表示的化合物、或其药理学上可接受的盐。[化学式1]〔式中,a1、a2各自独立地为氨基酸残基,m为0~3的整数,a3为在侧链上具有氨基或羧基的氨基酸残基,a4为氨基酸残基,n为0~3的整数,r1为与a3的侧链的氨基或羧基结合且具有能够与靶分子识别单元或其连接基团结合的官能团的基团、或者a3的侧链的氨基或羧基的氢原子,其中,r1可以形成碳原子数为3~10的杂环基,所述杂环基含有a3的侧链的氨基的氮原子作为成环原子之一,l为式(l1)表示的基团、或式(l2)表示的基团,[化学式2](式中,r3,r4,r5,r6各自独立地为氢原子、-ch2coor10基、或碳原子数为1~8的烃基,r10为氢原子或碳原子数为1~8的烃基,*为结合位点。其中,r3,r4,r5,r6之中,至少3个以上为-ch2cooh基。)[化学式3](式中,*为结合位点。)〕〔2〕化合物、或其药理学上可接受的盐,其是靶分子识别单元与上述〔1〕所述的化合物、或其药理学上可接受的盐结合而成的。〔3〕金属络合物、或其药理学上可接受的盐,其具有:选自由放射性金属及放射性原子标记金属组成的组中的1种金属、及与所述金属配位的〔1〕或〔2〕所述的化合物或其药理学上可接受的盐。〔4〕放射性药物的制备用药物,其包含上述〔1〕或〔2〕所述的化合物、或其药理学上可接受的盐。〔5〕上述〔1〕或〔2〕所述的化合物、或其药理学上可接受的盐的、用于制造放射性药物的用途。〔6〕放射性药物,其包含上述〔3〕所述的金属络合物、或其药理学上可接受的盐。〔7〕放射线治疗药,其包含上述〔3〕所述的金属络合物、或其药理学上可接受的盐。〔8〕放射性成像诊断药物,其包含上述〔3〕所述的金属络合物、或其药理学上可接受的盐。〔9〕上述〔1〕或〔2〕所述的化合物、或其药理学上可接受的盐,其中,所述化合物、或其药理学上可接受的盐用于放射性药物的制备。〔10〕上述〔3〕所述的金属络合物、或其药理学上可接受的盐的、用于制造放射性药物的用途。〔11〕上述〔3〕所述的金属络合物、或其药理学上可接受的盐的、用于放射线治疗的用途。〔12〕上述〔3〕所述的金属络合物、或其药理学上可接受的盐的、用于放射线成像诊断的用途。〔13〕放射线治疗方法,其施予上述〔3〕所述的金属络合物、或其药理学上可接受的盐。〔14〕放射线成像诊断方法,其施予上述〔3〕所述的金属络合物、或其药理学上可接受的盐。〔15〕试剂盒,其以单独的包装单元包含:上述〔1〕所述的化合物或其药理学上可接受的盐、或者靶分子识别单元与上述〔1〕所述的化合物、或其药理学上可接受的盐结合而成的化合物、或其药理学上可接受的盐;和含有选自由放射性金属及放射性原子标记金属组成的组中的1种金属的药物。发明效果根据本发明,可以提供能够获得放射性药物的化合物等,所述放射性药物能够利用包括具有较大原子半径的原子在内的多种原子进行标记,能够减少向肾脏的积聚。附图说明[图1]图1示出了111in-cdo3aet-fgk(boc)与bbmvs孵育的实验结果。[图2]图2示出了111in-cdota-bn-co-fgk(boc)溶液与bbmvs孵育的实验结果。[图3]图3示出了111in-do3a-bn-scn-mvk(bzo)与bbmvs孵育的实验结果。[图4]图4示出了111in-do3a-bn-co-fgk(boc)与bbmvs孵育的实验结果。[图5]图5示出了111in-cdo3aibu-fgk(boc)与bbmvs孵育的实验结果。[图6]图6示出了111in-cdo3aet-fgk-fab(来源于兔血清igg)与111in-do3a-eda-fab(来源于兔血清igg)的结果的比较。[图7]图7示出了111in-cdo3aibu-fgk-fab(来源于抗c-kitigg)、111in-dota-bn-scn-fab(来源于抗c-kitigg)、及111in-cdo3aet-fgk-fab(来源于抗c-kitigg)的结果的比较。[图8]图8示出了将111in-cdo3aet-fgk-fab(来源于兔血清igg)施予至小鼠后24小时为止排泄的尿液中放射活性的化学形态的分析结果。[图9]图9示出了将111in-cdota-bn-co-fgk-fab(来源于抗c-kitigg)施予至小鼠后6小时为止排泄的尿液中放射活性的化学形态的分析结果。[图10]图10示出了将111in-cdo3aet-fgk-fab(来源于兔血清igg)施予至小鼠后24小时为止排泄的尿液中放射活性的化学形态的分析结果。[图11]图11示出了将111in-cdo3aibu-fgk-fab(来源于兔血清igg)施予至小鼠后24小时为止排泄的尿液中放射活性的化学形态的分析结果。[图12]图12为将111in-do3a-bn-scn-mvk-fab(来源于抗c-kitigg)溶液、或111in-cdo3aet-fgk-fab(来源于抗c-kitigg)溶液施予至sy皮下肿瘤模型小鼠后2.5小时的spect/ct成像。具体实施方式[化合物等]<化合物(1)等>本发明的化合物、或其药理学上可接受的盐(下文中也简称为“化合物(1)等”)由下述式(1)表示。[化学式4]〔式中,a1、a2各自独立地为氨基酸残基,m为0~3的整数,a3为在侧链上具有氨基或羧基的氨基酸残基,a4为氨基酸残基,n为0~3的整数,r1为与a3的侧链的氨基或羧基结合且具有能够与靶分子识别单元或其连接基团结合的官能团的基团、或者a3的侧链的氨基或羧基的氢原子,其中,r1可以形成碳原子数为3~10的杂环基,所述杂环基含有a3的侧链的氨基的氮原子作为成环原子之一,l为式(l1)表示的基团、或式(l2)表示的基团,[化学式5](式中,r3、r4、r5、r6各自独立地为氢原子、-ch2coor10基、或碳原子数为1~8的烃基,r10为氢原子或碳原子数为1~8的烃基,*为结合位点。其中,r3、r4、r5、r6之中,至少3个以上为-ch2cooh基。)[化学式6](式中,*为结合位点。)〕根据本发明,可以提供能够获得放射性药物的化合物等,所述放射性药物能够利用铟-111进行标记,能够减少向肾脏的积聚。此外,由于本发明的放射性药物具有靶分子识别单元,因此其可以特异性地结合于靶位点,从而有效地在靶位点积聚。由于这样的性质,本发明的放射性药物能够在放射线治疗中特异性地积聚于肿瘤部位,在放射线成像诊断中提高其灵敏度、精度。能够获得本发明效果的原因尚不确定,但可以认为如下。可以认为,如果在施予放射性药物后,该药物被肾细胞摄入时,能够高效地使尿液排泄性的放射性代谢物游离,则可以减少向肾脏的放射活性的积聚。因此,为了在放射性药物被肾细胞摄入时,能够高效地使包纳有放射性标记元素的螯合配体部位游离,从而在多肽与螯合配体部位之间导入肾刷状缘膜酶的底物序列。可推测通过上述方式,在被摄入肾细胞之前,使多肽和螯合配体部位从多肽游离,抑制放射性物质进入肾脏,从而能够自施予早期起减少向肾脏的积聚。为了能实现利用铟-111的标记,研究了导入式(l1)或式(l2)表示的基团作为螯合配体部位。这种情况下,考虑了如例如专利文献1中记载的化合物那样,导入硫代脲结构作为将螯合药物部位与多肽部位连接的结构。然而,发明人实验表明,使用具有式(l1)或式(l2)表示的基团的化合物作为螯合药物时,若导入具有该硫代脲结构的连接基团,则不会发生基于肾刷状缘膜酶的分解。针对这一点,发现通过如式(1)表示的化合物那样导入特定结构的连接基团,从而发生基于肾刷状缘膜酶的分解,能够减少向肾脏的放射活性的积聚。就本发明的化合物(1)等而言,从自施予早期起减少向肾脏的积聚的观点考虑,式(1)中,a1至a4的氨基酸序列(其中,n=0的情况下,为a1至a3的氨基酸序列)优选为与肾刷状缘膜酶的底物序列的一部分相同的序列。从自施予早期起减少向肾脏的积聚的观点考虑,a1优选为苯丙氨酸、甲硫氨酸、缬氨酸、亮氨酸、异亮氨酸、脯氨酸、酪氨酸、甘氨酸、丙氨酸或色氨酸的残基,更优选为苯丙氨酸、甘氨酸、丙氨酸或甲硫氨酸的残基,从使自施予早期起减少向肾脏的积聚的效果更显著的观点考虑,进一步优选为苯丙氨酸的残基。从自施予早期起减少向肾脏的积聚的观点考虑,a2优选为甘氨酸、苯丙氨酸、甲硫氨酸、缬氨酸、亮氨酸、异亮氨酸、脯氨酸、酪氨酸、丙氨酸或色氨酸的残基,更优选为甘氨酸、苯丙氨酸、丙氨酸、缬氨酸或异亮氨酸的残基,从使自施予早期起减少向肾脏的积聚的效果更显著的观点考虑,进一步优选为甘氨酸的残基。需要说明的是,m为0~3的整数,优选为1。从在氨基酸序列的侧链上导入能够与多肽或其连接基团结合的官能团的观点考虑,a3为在侧链具有氨基或羧基的氨基酸残基,优选为赖氨酸、鸟氨酸、精氨酸、天冬氨酸或谷氨酸的残基,更优选为赖氨酸、鸟氨酸或精氨酸的残基,进一步优选为赖氨酸的残基。需要说明的是,作为a4,可具有其他氨基酸残基。作为a4,可使用任意的氨基酸。n为0~3的整数,优选为0。就r1而言,为具有能够与靶分子识别单元或其连接基团结合的官能团的基团、或a2的侧链的氨基或羧基的氢原子,且与a3的侧链的氨基或羧基结合。其中,r1可以形成碳原子数为3~10的杂环基,所述杂环基含有a3的侧链的氨基的氮原子作为成环原子之一。r1作为间隔基团发挥功能,可以介由官能团将多肽等靶分子识别单元与本发明化合物结合。r1与a3的侧链的氨基或羧基结合,由此可以不对氨基酸序列末端进行化学修饰,而将本发明的化合物与多肽结合。r1可以与侧链的氨基的氮原子结合,也可以与侧链的羧基形成酯键。r1的能够与靶分子识别单元或其连接基团结合的官能团没有特别限定,可举出例如羧基或其活性酯;马来酰亚胺基、丙烯酰基等具有c=c键的基团;选自由氨基甲酰基、异硫氰酸酯基及氨基组成的组中的至少1种官能团(以下也称为“官能团a”)。作为羧基的活性酯,可举出氯乙酰基、溴乙酰基、碘乙酰基等。其中,官能团a优选为具有c=c键的基团、氨基甲酰基。r1的碳原子总数没有特别限定,例如优选为1以上,更优选为2以上,进一步优选为3以上,并且,优选为20以下,更优选为10以下,进一步优选为8以下。作为r1,可举出例如具有官能团a的碳原子总数为2~20的酰基、具有官能团a的碳原子总数为2~20的烷基、具有官能团a的碳原子总数为2~20的烷基氨基甲酰基、具有官能团a的碳原子总数为2~20的烷基硫代氨基甲酰基。r1形成杂环基的情况下,杂环基优选为马来酰亚胺基。r1形成杂环基的情况下,杂环基的碳原子数优选为3~10,更优选为3~5,进一步优选为4或5。r1可以是a3的侧链的氨基或羧基的氢原子。也即,a3的氨基或羧基可以不进行修饰。r1之中,优选含有a3的侧链的氨基的氮原子作为成环原子之一的碳原子数为3~10的杂环基,更优选含有a3的侧链的氨基的氮原子作为成环原子之一的马来酰亚胺基。l为式(l1)表示的基团、或者式(l2)表示的基团,[化学式7][化学式8]关于式(l1)表示的基团,r3、r4、r5、r6之中,优选3个以上且4个以下为-ch2cooh基,更优选3个为-ch2cooh基。作为-ch2coor10基,r10为氢原子或碳原子数为1~8的烃基。r3、r4、r5、r6之中,-ch2cooh基以外的基团优选为碳原子数为1~8的烃基,更优选为碳原子数为1~4的烃基。从减少向肾脏的积聚的观点考虑,r3,r4,r5,r6之中,至少1个基团优选为碳原子数为3~8的烃基,更优选为碳原子数为4~6的烃基。该烃基优选为脂肪族烃基,更优选为支链脂肪族烃基。作为该烃基,可举出例如甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基。其中,从减少向肾脏的积聚的观点考虑,优选乙基、正丁基、仲丁基、异丁基,更优选正丁基、仲丁基、异丁基,进一步优选异丁基。其中,l优选为式(l1)表示的基团:[化学式9](式中,r3,r4,r5,r6为-ch2cooh基、或碳原子数为1~8的烃基,*为结合位点。其中,r3,r4,r5,r6之中,3个为-ch2cooh基。)。作为l,优选为选自由式(l1-1)、式(l1-2)、式(l1-3)、式(l1-4)、式(l1-5)、式(l1-6)、式(l1-7)、式(l1-8)、式(l1-9)及式(l2)组成的组中的至少1种。[化学式10][化学式11](以上,式中,*为结合位点。)以上的本发明的化合物(1)中,优选下述式(1a)表示的化合物。[化学式12]〔式中,l为式(l1)表示的基团、或式(l2)表示的基团,[化学式13](式中,r3、r4、r5、r6各自独立地为氢原子、-ch2coor10基、或碳原子数为1~8的烃基,r10为氢原子或碳原子数为1~8的烃基,*为结合位点。其中,r3、r4、r5、r6之中,至少3个以上为-ch2cooh基。)[化学式14](式中,*为结合位点。),r7为氢原子或甲基,r8,r9各自独立地为氢原子、或者具有官能团a的碳原子总数为2~20的酰基、具有官能团a的碳原子总数为2~20的烷基、具有官能团a的碳原子总数为2~20的烷基氨基甲酰基、或具有官能团a的碳原子总数为2~20的烷基硫代氨基甲酰基。其中,r8及r9可以形成含有相邻氮原子的杂环,这种情况下,由下式:[化学式15]表示的基团为由下式:[化学式16]表示的基团。〕r8,r9优选为具有官能团a的碳原子总数为2~20的酰基,更优选为具有氨基甲酰基的碳原子总数为3~6的酰基。作为具有氨基甲酰基的碳原子总数为3~6的酰基,可举出例如式:-c(=o)(ch2)a(=o)nh2〔式中,a为1~4的整数。〕表示的基团。就上述式(1a)表示的化合物而言,优选的是,式中,l为式(l1)表示的基团:[化学式17](式中,r3、r4、r5、r6各自独立地为-ch2cooh基、或碳原子数为1~8的烃基,*为结合位点。其中,r3、r4、r5、r6之中,3个为-ch2cooh基,1个为碳原子数为1~8的烃基。),更优选的是,式中,l为式(l1)表示的基团:[化学式18](式中,r3、r4、r5、r6各自独立地为-ch2cooh基、乙基、或丁基,*为结合位点。其中,r3、r4、r5、r6之中,3个为-ch2cooh基,1个为乙基、或丁基。),进一步优选的是,式中,l为式(l1)表示的基团:[化学式19](式中,r3、r4、r5、r6各自独立地为-ch2cooh基、异丁基,*为结合位点。其中,r3、r4、r5、r6之中,3个为-ch2cooh基,1个为异丁基。)。作为上述本发明的化合物(1)的优选具体例,可举出下述的化合物1-1~化合物1-6。[化学式20][化学式21][化学式22][化学式23]本发明的化合物(1)等可以是上述化合物的药理学上可接受的盐。作为药理学上可接受的盐,可举出例如酸加成盐、碱加成盐。作为酸加成盐,可以是无机酸盐或有机酸盐中的任何。作为无机酸盐,可举出例如盐酸盐、氢溴酸盐、硫酸盐、氢碘酸盐、硝酸盐、磷酸盐等。作为有机酸盐,可举出例如柠檬酸盐、草酸盐、乙酸盐、甲酸盐、丙酸盐、苯甲酸盐、三氟乙酸盐、马来酸盐、酒石酸盐、甲磺酸盐、苯磺酸盐、对甲苯磺酸盐等。作为碱加成盐,可以是无机碱盐或有机碱盐中的任何。作为无机碱盐,可举出例如钠盐、钾盐、钙盐、镁盐、铵盐等。作为有机碱盐,可举出例如三乙基铵盐、三乙醇铵盐、吡啶鎓盐、二异丙基铵盐。<化合物(2)等>本发明的化合物(2)等是靶分子识别单元与化合物(1)或其药理学上可接受的盐结合而成的化合物、或其药理学上可接受的盐。靶分子识别单元可以介由连接基团而与化合物(1)、或其药理学上可接受的盐结合,也可以直接与化合物(1)、或其药理学上可接受的盐结合。作为连接基团,可举出衍生自2-亚氨基硫杂环戊烷(iminothiolane)的亚氨基硫醇。〔靶分子识别单元〕“靶分子识别单元”是指:能够在生物体内与靶分子结合等的并能识别靶分子的分子、取代基、官能团或原子团。作为靶分子识别单元,可举出多肽、以及与靶分子结合的配体。多肽通常是与靶分子结合的多肽,并且优选为与靶分子特异性结合的多肽。特异性结合是指,与靶分子结合,但对于靶分子以外的分子则为不结合、或弱结合。靶分子是指,介由放射性药物而成为诊断对象的靶位点,例如存在于组织、细胞中的分子,优选为特异性表达的分子。“特异性表达”是指,在靶位点表达,但在靶位点以外的位点则为不表达、或低表达。作为靶分子识别单元,可举出例如:与在伴随炎症、肿瘤细胞浸润等的组织构建中被发现高表达的蛋白质、或在肿瘤细胞中特异性表达的蛋白质结合的配体、以及抗体及抗体的抗原结合区片段。作为抗体,可举出例如抗cd25抗体、抗cd20抗体等单克隆抗体。作为抗体的抗原结合区片段,可举出例如fab片段(以下简称为“fab”)、f(ab')2片段、f(ab)2片段、可变区片段(以下也称为“fv片段”)。fab片段是指,通过木瓜蛋白酶分解抗体产生的n末端侧的产物、及具有与其相同的结构域结构的片段。f(ab')2片段是指,通过还原抗体的f(ab')2的铰链区的二硫键而得到的片段、及具有与其相同的结构域结构的片段。f(ab)2片段是指,将两分子的fab片段通过二硫键彼此结合而得到的二聚体。fv片段是指,抗体的片段,并且是具有与抗原的结合活性的最小片段。作为抗体的抗原结合区片段,更具体而言,可举出与在特定癌细胞中特异性表达的蛋白质相对应的抗体、及其fab片段或fv片段。作为其他靶分子识别单元,可举出:对在癌的新生血管中高水平表达的整联蛋白具有亲和性的环状五肽,例如环-arg-gly-asp-d-phe-lys(以下也称为“c(rgdfk)”)。此外,可举出:对成骨癌(骨转移)中大量存在的羟基磷灰石具有亲和性的双膦酸、低聚天冬氨酸、低聚谷氨酸、对存在于巨噬细胞表面的趋化因子受体具有亲和性的肽即fmet-leu-phe(fmlp)、与在癌细胞中表达的叶酸受体结合的叶酸及其衍生物等。需要说明的是,靶分子识别单元不限于这些示例出的多肽,只要其为与靶分子结合的多肽即可使用任何。就靶分子识别单元而言,例如可以使用2-亚氨基硫杂环戊烷等硫醇化试剂来导入与化合物的官能团反应的连接基团并使其结合。该连接基团向fab片段的导入,可以通过使所述硫醇化试剂在ph7-9的条件下反应,对fab截面上的氨基附加巯基来实现。作为靶分子识别单元,可以使用例如具有asn-urea-lys位点或glu-urea-lys位点的配体。通过该配体,选择性地结合在前列腺癌中表达显著升高的前列腺特异性膜抗原(prostatespecificmembraneantigen)的受体。asn-urea-lys位点是由下式表示的位点:[化学式24]〔式中,*为结合位点。〕。glu-urea-lys位点是由下式表示的位点:[化学式25]〔式中,*为结合位点。〕。除上述以外,可举出例如下述方法:使导入特定的官能团f1后的上述的多肽、除此以外还有与靶分子结合的配体与在伴随炎症、肿瘤细胞浸润等的组织构建中被发现高表达的蛋白质、或在肿瘤细胞中特异性表达的蛋白质等靶分子结合,作为靶分子识别单元,施予具有与官能团f1反应并成键的官能团f2的化合物(2)等,来识别靶分子[chemicalsocietyreviews45:6409-6658,2016,chemicalsocietyreviews42:5131-5142,2013]。作为官能团f1,可举出例如下述的式(f1-1)、式(f1-2)、或式(f1-3)表示的基团。[化学式26]〔式中,*为结合位点。〕作为官能团f2,可举出例如下述的式(f2-1)、式(f2-2)、式(f2-3)、式(f2-4)、或式(f2-5)表示的基团。[化学式27]〔式中,*为结合位点。〕使用本发明的化合物(2)等,可提供用于制备包含该化合物的放射性药物的药物。除该化合物外,放射性药物的制备用药物可还含有水性缓冲液等ph调节剂、抗坏血酸、对氨基苯甲酸等稳定剂。作为本发明的化合物(2)等,可举出例如以下的式(2)表示的化合物、或其药理学上可接受的盐。[化学式28]〔式中,a1、a2、m、a3、a4、n、r1、l与式(1)相同,l1为将r1与p1连接的连接基团,p为0或1,p1为靶分子识别单元。〕l1经由能与r1的连接基团连接的官能团成键,与靶分子识别单元也成键。l1优选为衍生自2-亚氨基硫杂环戊烷的亚氨基硫醇等。p优选为1。p1为例如上述的靶分子识别单元,优选为多肽、以及与靶分子结合的配体、或者式(f2-1)、式(f2-2)、式(f2-3)、式(f2-4)或式(f2-5)表示的官能团f2。作为上述本发明的化合物(2)的优选具体例,可举出下述的化合物2-1~化合物2-4。其中,下述的式中fab表示fab片段部位。[化学式29][化学式30][化学式31][化学式32]<金属络合物(3)等>本发明的金属络合物、或其药理学上可接受的盐(以下也称为“金属络合物(3)等”)具有:选自由放射性金属及放射性原子标记金属组成的组中的1种金属、及与所述金属配位的本发明的化合物或其药理学上可接受的盐。就包含本发明的金属络合物(3)等的放射性药物而言,除金属络合物(3)等之外还可以含有未反应物、杂质,也可以为包含在制造后通过高效液相色谱(hplc)法等纯化而得的金属络合物(3)等的物质。术语“络合物”是指配体以金属和类金属元素的原子或离子为中心进行配位而得到的物质,也称为配位化合物。配位是指配体与中心的金属形成配位键并且在中心金属的周围排列。络合物由配体和金属之间的配位键形成。也将由配体和金属进行的络合物的形成称为络合。配位键是指,形成一个键的两个价电子仅由一个原子提供的键。〔金属〕作为金属,可举出例如111in、223ra、67ga、68ga、44sc、90y、177lu、225ac、212bi、213bi、212pb、227th、64cu、67cu。金属优选为选自由111in、223ra、67ga、68ga、90y、177lu、225ac、212bi、213bi、212pb、及227th组成的组中的至少1种,更优选为选自由111in、90y、177lu、225ac、212bi、213bi及212pb组成的组中的至少1种,进一步优选为111in、90y、177lu及225ac。金属不限于这些具体例,只要其具有适用于使用了放射性药物的诊断等目的的放射线、放射线量、半衰期则均可使用。从减少放射线成像诊断中对正常的组织、细胞的影响的观点考虑,优选使用半衰期短的金属放射性同位素。金属络合物(3)等的制造可以通过使用与靶分子识别单元结合的所述化合物作为配体,并在体外与金属放射性同位素络合来实现。络合可以通过利用以往已知的络合反应的简便操作来实现。作为本发明的金属络合物(3),例如,以化合物(1)的l为式(l1-4)、式(l1-5)、式(l1-9)或式(l2)表示的基团的情况为例,可举出以下的式(3-1)表示的金属络合物。[化学式33]〔式中,a1、a2、m、a3、a4,n、r1与式(1)相同,l’为式(l1’-4)、式(l1’-5)、式(l1’-9)、或式(l2’)表示的基团:[化学式34](式中,m为111in、223ra、67ga、68ga、44sc、90y、177lu、225ac、212bi、213bi、212pb、227th、64cu、或67cu。)〕作为本发明的金属络合物(3),例如,以化合物(2)的l为式(l1-4)、式(l1-5)、式(l1-9)、或式(l2)表示的基团的情况为例,可举出以下的式(3-2)表示的金属络合物。[化学式35]〔式中,a1,a2,m,a3,a4,n,r1与式(1)相同,l1,p,p1与式(2)相同,l’为式(l1’-4)、式(l1’-5)、式(l1’-9)或式(l2’)表示的基团:[化学式36](式中,m为111in、223ra、67ga、68ga、44sc、90y、177lu、225ac、212bi、213bi、212pb、227th、64cu、或67cu。)。〕本发明的放射性药物含有上述放射性标记多肽作为有效成分,此外还可以制备为根据需要而含有1种或2种以上医药上可接受的载体(医药用载体)的医药组合物。作为医药用载体,可例举水性缓冲液、酸、以及碱等ph调节剂、抗坏血酸或对氨基苯甲酸等稳定剂、d-甘露醇等赋形剂、等渗剂、以及防腐剂等。此外,可以添加用于改善放射化学性纯度的柠檬酸、酒石酸、丙二酸、葡糖酸钠、葡庚糖酸钠等化合物。本发明的放射性药物可以以水溶液的形式、冷冻溶液的形式或冷冻干燥产品中的任何形式提供。本发明的试剂盒以单独的包装单元包含上述化合物、和含有上述金属的药物。本发明的试剂盒可举出:例如,以单独的包装单元包含化合物(1)等含有靶分子识别单元的药物、以及含有选自由放射性金属及放射性原子标记金属组成的组中的1种金属的药物的试剂盒;以单独的包装单元包含靶分子识别单元与化合物(1)等结合而成的化合物(2)等、以及含有选自由放射性金属及放射性原子标记金属组成的组中的1种金属的药物的试剂盒。试剂盒中包含的化合物和药物均可根据需要而含有如上所述的1种或2种以上医药上可接受的载体(医药用载体)。[制造方法]本发明的化合物(1)等、以及靶分子识别单元与该化合物(1)等结合而成的化合物(2)等可以通过已知的方法合成,可以通过例如本说明书的实施例中记载的方法来制造。本发明的金属络合物(3)等可以通过使用化合物(2)等作为配体、并在体外与金属放射性金属或放射性原子标记金属络合来制造。络合可以使用已知的方法进行。[使用容量]本发明的金属络合物等可以用作例如放射线治疗或放射线成像诊断中使用的放射性药物。本发明的金属络合物等可以用于通过将其有效量施予至包括人在内的哺乳动物来抑制癌症的放射线治疗。作为抗癌剂使用的情况下,具有包括例如防止癌的发生、或转移·着床、复发的预防作用、以及抑制癌细胞的增殖、或通过使癌缩小来阻止癌症的进行、或使症状改善的治疗作用这两者的最宽泛的含义,在任何情况下均不作限定性解释。可用作放射线治疗药的、选自由放射性金属及放射性原子标记金属组成的组中的1种金属可举出例如发射α射线的核素、发射β射线的核素、发射γ射线的核素、发射正电子的核素。其中,放射线治疗的用途中,优选发射β射线的核素(即,释放β射线的核素)。作为放射线成像诊断,可举出例如单光子放射断层扫描(singlephotonemissioncomputedtomography,下文中也简称为“spect”)、正电子放射断层扫描(positronemissiontomography,下文中也简称为“pet”)等。作为诊断,没有特别限定,可以用于肿瘤、炎症、传染病、心血管疾病、脑/中枢系统疾病等各种疾病和器官/组织的放射线成像诊断等,优选用于癌症的放射线成像诊断。通过根据作为诊断对象的靶标的特性来选择靶分子识别单元,可实现多种类多靶点的诊断、治疗,本发明的放射性药物可以在诊断领域作为放射性成像诊断药物而广泛地使用。作为本发明的放射性药物的施予途径,可举出例如静脉内施予或动脉内施予等非经口施予、经口施予,优选静脉内施予。施予途径不限于这些途径,只要该途径可以在放射性药物的施予后有效地表达其作用,则可以使用任何途径。本发明的放射性药物的放射活性强度为通过施予本药物可以达到目的的强度,并且,只要是使受试者的放射线暴露尽可能低的临床施予量即可。放射性强度可以参考使用放射性药物的常规诊断方法、治疗方法中所使用的放射活性强度来确定。就其施予量而言,可以考虑患者的年龄、体重、适合的放射线成像装置、以及对象疾病的状态等各条件,来确定可成像的放射能及施予量。在以人为对象的情况下,放射性药物中的放射能量如下所述。通常,假定其用于放射线治疗,并且诊断药物的施予量没有特别限定,例如,作为放射性金属(例如111in)的放射能量,为1.0mbq/kg~3.0mbq/kg。如上所述,根据本发明,可以提供能够获得放射性药物的化合物,所述放射性药物能够自施予早期起减少向肾脏的积聚。实施例以下说明的本发明的实施例仅用于示例,并不限制本发明的技术范围。需要说明的是,以下实验经千叶大学动物伦理委员会批准后进行。在以下实施例和比较例中,对于取代基、化合物、以及有机溶剂使用下述缩写。fmoc:芴基甲氧基羰基boc:叔丁氧基羰基thf:四氢呋喃nmm:n-甲基吗啉dmf:二甲基甲酰胺tfa:三氟乙酸基dcc:n,n’-二环己基碳二亚胺trt(2-cl):2-氯三苯甲基cl-trt(2-cl)resin:2-氯三苯甲基氯树脂(2-chlorotritylchlorideresin)fmoc-lys(dde)-oh:n-α-(9-芴基甲氧基羰基)-n-ε-[1-(4,4-二甲基-2,6-二氧代亚环己基)乙基]-l-赖氨酸tfa:三氟乙酸mecn:乙腈dipea:n,n-二异丙基乙胺dic:n,n'-二异丙基碳二亚胺hobt:1-羟基苯并三唑nmcm:n-甲氧基羰基马来酰亚胺edta:乙二胺四乙酸dps:2,2'-联吡啶二硫化物etoh:乙醇fbs:胎牛血清d-pbs:杜氏磷酸盐缓冲生理盐水egta:乙二醇醚二胺四乙酸[测定方法、实验动物]在下述实施例和比较例中,各种物性等的测定方法通过以下方法进行。〔nmr(核磁共振)〕基于1h-nmr的分析使用jeolecs-400光谱仪(日本电子株式会社)。〔esi-ms(电喷雾电离质谱)〕基于esi-ms的分析使用hplc1200系列-6130四极杆lc/ms质谱仪(agilenttechnologies株式会社)。〔薄层色谱(tlc)〕基于薄层色谱(tlc)的分析使用二氧化硅板(tlcaluminiumsheetssilicagel60rp-18f254s,merck株式会社),将用0.1m乙酸铵水溶液:甲醇=1:1的展开溶液展开10cm而得的二氧化硅板每隔5mm进行切割,并通过自然伽马测井仪(wizard3,perkinelmer公司)测定各级分的放射活性。〔乙酸纤维素膜电泳(cae)〕在乙酸纤维素膜电泳(以下也称为“cae”)中,电泳膜使用切成11cm×1cm尺寸的乙酸纤维素膜(advantecseleca-v,东洋滤纸株式会社),缓冲液使用(veronal缓冲液,ph8.6,i=0.06)或溶剂2(20mmp.b.(ph6.0)),并通过恒定电流(1ma/cm)进行电泳。将电泳后的乙酸纤维素膜每隔5mm进行切割,并且通过自动井伽马系统测定各级分的放射活性。〔反相高效液相色谱(rp-hplc)和分子筛高效液相色谱(se-hplc)〕(分析)基于反相高效液相色谱(以下也称为“rp-hplc”)的分析使用l-7405(株式会社日立制作所)作为uv检测器、使用l-7100(株式会社日立制作所)作为送液泵、并使用cosmosil5c18-ar-300柱(4.6×150mm,nacalaitesque株式会社)作为分析用色谱柱。使用0.1%(v/v)tfa/h2o(a相)和0.1%(v/v)tfa/mecn(b相)作为流动相,通过线性梯度法在0-20分钟内从a相95%(v/v)、b相5%(v/v)变化为a相70%(v/v)、b相30%(v/v),在20-40分钟内从a相70%(v/v)、b相30%(v/v)变化为a相0%(v/v)、b相100%(v/v),以1.0ml/分钟的流速洗脱。(分离)基于rp-hplc的分离使用连接有cadenza5cd-c18保护柱(10×8mm,intact株式会社)的cadenza5cd-c18柱(20×150mm,intact株式会社)。使用0.1%(v/v)tfa/h2o(a相)和0.1%(v/v)tfa/mecn(b相)作为流动相,通过线性梯度法在0-30分钟内从a相90%(v/v)、b相10%(v/v)变化为a相20%(v/v)、b相80%(v/v),在30-40分钟内从a相20%(v/v)、b相80%(v/v)变化为a相0%(v/v)、b相100%(v/v),以5.0ml/分钟的流速洗脱。对于基于分子筛hplc(下文中也称为“se-hplc”)的分析,将cosmosil5diol-300ii保护柱(7.5×50mm,nacalaitesque株式会社)连接到cosmosil5diol-300ii(7.5×600mm,nacalaitesque株式会社)上,并将0.1m磷酸缓冲液(ph6.8)用作流动相,以1.0ml/分钟的流速洗脱。通过rp-hplc测定洗脱液在254nm处的吸光度,通过se-hplc测定洗脱液在280nm处的吸光度,对于67ga标记化合物的分析通过在线连接γ射线检测器gabistar(raytest公司),或使洗脱液通过馏分收集器(frac-920,gehealthcarejapan株式会社)以0.5分钟间隔进行分离,然后利用自动井伽马系统测定放射活性来进行分析。〔试剂〕下述合成例中的“do3a-eda(mal)”为下式表示的化合物,使用了macrocyclics公司制的商品名“b-272”。[化学式37]〔实验动物〕动物实验使用了6周龄的雄性ddy系spf小鼠、及雄性的balb/c-nu/nu小鼠(japanslc株式会社)。[cdo3aet-fgk,cdo3aet-fgk(mal),cdo3aet-fgk(boc)的合成]合成例l1:化合物(a13)的合成[化学式38](a)氯甲酸异丁酯,n-甲基吗啉.thf,dmf;98%(b)4nhcl/acoet;99.8%(c)dcc,et3n,thf,acoet;91.2%(d)i.25%nh3aq,ii.hatu,hoat,diea54.2%;(e)4nhcl/acoet98.4%;(f)碘乙烷,k2co3,mecn;(g)i.0.95mbh3/thf,ii.浓盐酸;(h)溴乙酸叔丁酯,k2co3,mecn,dmf;(i)pd(oac)2,1,2-双(二苯基膦基)乙烷,et3n,co,bnoh,dmf;(j)10%pd/c,meoh合成例l1(a):化合物(a3)的合成将化合物(a1)(7.97g、20.4mmol)溶解于thf40ml中并冷却至-15℃后,在氩气气氛下,依次添加n-甲基吗啉(nmm,3.36ml、30.5mmol)及氯甲酸异丁酯(4.01ml、30.5mmol)。搅拌5分钟后,滴入化合物(a2)(5.87g、30.5mmol)和nmm(3.36ml、30.5mmol)的dmf溶液24ml,在冷却下搅拌30分钟,于室温搅拌1小时。减压蒸馏除去溶剂后,将残余物用乙酸乙酯100ml及5质量%碳酸氢钠水溶液100ml溶解,用5质量%碳酸氢钠水溶液(50ml×3)、5质量%柠檬酸水溶液(50ml×3)洗涤。向有机层加入无水硫酸镁并进行干燥,然后除去溶剂,将得到的晶体减压干燥,由此得到淡黄色晶体形态的化合物(a3)(10.6g、20.0mmol、收率98.0%)。1hnmr(cdcl3):δ1.42[9h,s,boc],2.96-3.03[2h,m,chch2],3.39-3.51[4h,ch2ch2],4.22-4.24[1h,dd,nhch],4.85,6.42,7.74[3h,t,nh],6.93-6.95[2h,d,cch],7.62-7.66[2h,d,icch].esi-ms(m+na)+:m/z552.07.found552.09.合成例l1(b):化合物(a4)将化合物(a3)(10.6g、20.0mmol)溶解于4n盐酸/乙酸乙酯50ml中,于室温搅拌3小时。减压蒸馏除去溶剂,减压干燥,由此得到淡黄色晶体形态的化合物(a4)(8.08g、20.0mmol、收率99.8%)。1hnmr(cdcl3):δ3.42-3.55[6h,overlapped,ch2],3.86-3.88[1h,dd,ch2ch],7.04-7.06[2h,d,cch],7.62-7.64[2h,d,icch].esi-ms(m+na)+:m/z452.02.found452.03.合成例l1(c):化合物(a6)将化合物(a5)(4.45g、19.1mmol)溶解于thf70ml中,将溶液冰冷却后,在氩气气氛下滴入dcc(4.30g、20.8mmol)的thf溶液20ml。滴入完成后,于室温搅拌1小时,过滤反应液,由此除去二环己基脲(以下也称为“dc-urea”),将滤液作为化合物(a5)的酸酐溶液用于下一反应。将化合物(a4)(8.08g、17.4mmol)悬浮于乙酸乙酯80ml中,加入et3n(3.63ml、26.0mmol)并冷却,搅拌10分钟后,在氩气气氛下,滴入之前制备的化合物(a5)的酸酐溶液。滴入完成后,于室温搅拌1小时,将反应液用5质量%柠檬酸水溶液(50ml×3)洗涤。向有机层加入无水硫酸镁并进行干燥,然后减压蒸馏除去溶剂,将得到的残余物利用以乙酸乙酯作为洗脱溶剂的硅胶柱色谱进行纯化,由此得到淡黄色晶体形态的化合物(a6)(10.2g、15.8mmol、收率91.2%)。1hnmr(cdcl3):δ1.37[9h,s,boc],3.00-3.19[2h,m,chch2],3.42-3.52[5h,overlapped,nch2,nhch2],3.77-4.03[2h,m,coch2],4.11-4.16[1h,dd,nch2],4.60-4.62[1h,t,ch2ch],6.96-7.01[2h,d,cch],7.58-7.60[2h,d,icch].esi-ms(m+na)+:m/z667.09.found667.02.合成例l1(d):化合物(a7)将化合物(a6)(3.52g、5.46mmol)溶解于25质量%氨水50ml中,于室温搅拌3小时。减压蒸馏除去溶剂后,减压干燥,将得到的油状物溶解于dmf90ml中,将所得溶液作为a液。将o-(7-氮杂-1h-苯并三唑-1-基)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu,3.12g、8.21mmol)溶解于dmf90ml中,将所得溶液作为b液。在dmf1600ml中溶解diea(3.81ml、21.9mmol)、1-羟基-7-氮杂苯并三唑(1.12g、8.22mmol),将a液及b液使用注射器泵以1.2ml/h同时以低速滴入,滴入结束后搅拌24小时。减压蒸馏除去溶剂,将得到的残余物用乙酸乙酯及己烷洗涤,由此得到白色晶体形态的化合物(a7)(1.57g、2.96mmol、收率54.2%)。1hnmr(cd3od):δ1.46[9h,s,boc],2.82-2.89[1h,m,ch2],2.91-2.96[2h,m,ch2],3.03-3.14[1h,m,ch2],3.56-3.64[2h,m,ch2],3.82-4.02[3h,m,ch2],4.10-4.18[1h,m,ch2],4.46-4.50[1h,dd,ch],7.02-7.04[2h,d,ch],7.60-7.62[2h,d,ch].esi-ms(m+na)+:m/z553.09.found553.10.合成例l1(e):化合物(a8)将化合物(a7)(1.57g、2.96mmol)悬浮于4n盐酸/乙酸乙酯40ml中,于室温搅拌4小时。减压蒸馏除去溶剂,将得到的晶体用己烷洗涤,减压干燥,由此得到白色晶体形态的化合物(a8)(1.36g、2.92mmol、收率98.4%)。1hnmr(cdcl3):δ2.83-2.89[1h,m,ch2],2.96[1h,s,ch2],3.12-3.19[1h,m,ch2],3.40-3.52[2h,m,ch2],3.62-3.59[2h,m,ch2],4.07-4.13[1h,m,ch2],4.19-4.42[1h,m,ch2],4.60-4.62[1h,dd,ch],6.11[1h,s,nh],6.54[1h,s,nh],6.96-7.01[2h,d,ch],7.58-7.60[2h,d,ch].esi-ms(m+h)+:m/z431.06.found431.03.合成例l1(f):化合物(a9)将化合物(a8)(880mg、2.04mmol)悬浮于dmf13.5ml中,进而加入碳酸钾(424mg、3.06mmol)使其悬浮。将溶液冰冷却,在氩气气氛下滴入碘乙烷(327μl、4.08mmol)。滴入完成后,于80℃搅拌四天。减压蒸馏除去溶剂,将得到的残余物悬浮于乙酸乙酯中并进行过滤。将滤液用5质量%碳酸氢钠水溶液洗涤。向有机层中加入硫酸钠并干燥后,减压蒸馏除去溶剂,将得到的残余物利用使用了氯仿和甲醇的flash色谱系统进行生成,由此得到白色晶体形态的化合物(a9)(340mg、0.742mmol、收率36.3%)。1hnmr(cdcl3):δ1.10[3h,t,ch3],2.76-2.84[2h,q,ch2],2.85-2.89[1h,m,ch2],3.08-3.25[6h,m,ch2],3.37-3.43[1h,m,ch2],3.53-3.57[1h,m,ch2],3.69-3.75[1h,m,ch2],4.61-4.66[1h,dd,ch],6.59[1h,s,nh],6.84[1h,s,nh],6.97-6.99[2h,d,ch],7.16[1h,s,nh],7.58-7.60[2h,d,ch].esi-ms(m+h)+:m/z459.09.found459.17.合成例l1(g):化合物(a10)将化合物(a9)(340mg、0.742mmol)悬浮于thf1.4ml中,将溶液冰冷却后,在氩气气氛下,缓缓加入0.95m的硼烷-thf络合物/thf溶液13.5ml,搅拌1小时后,回流24小时。将溶液冰冷却,加入甲醇13.5ml后,搅拌1小时,减压蒸馏除去溶剂。然后,向残余物中再次加入甲醇13.5ml,减压蒸馏除去溶剂。向残余物中加入浓盐酸13.5ml,于室温搅拌24小时后,回流1小时。将溶液冰冷却,加入12.5n氢氧化钠水溶液使其成为碱性后,用氯仿萃取。向有机层中加入硫酸钠并干燥后,减压蒸馏除去溶剂,将得到的残余物利用以氯仿:甲醇:25质量%氨水(10:1:0.1)作为洗脱溶剂的flash色谱系统进行纯化,由此得到黄色油状物形态的化合物(a10)(163mg、0.391mmol、收率52.7%)。合成例l1(h):化合物(a11)将化合物(a10)(197mg、0.401mmol)溶解于乙腈2.75ml和dmf0.55ml的混合液中,进而加入碳酸钾(229mg、1.65mmol)使其悬浮。将溶液冰冷却,在氩气气氛下,滴入溴乙酸叔丁酯(229μl、1.40mmol)。滴入完成后,于室温搅拌48小时,过滤悬浮液后,从滤液中减压蒸馏除去溶剂。将残余物溶解于少量的氯仿中,移至厚度为1mm的分离用tlc,以氯仿:甲醇=8:1作为展开溶剂,由此进行纯化,得到红褐色油状物形态的化合物(a11)(306mg、0.403mmol、100%)。1hnmr(cdcl3):δ0.98-1.03[3h,t,ch3],1.41-1.48[27h,m,tbu],1.90-4.90[27h,m,ch2,dota],6.89-7.07[2h,m,ch],7.50-7.61[2h,m,ch].esi-ms(m+h)+:m/z759.36.found759.24.合成例l1(i):化合物(a12)将化合物(a11)(306mg、0.403mmol)悬浮于dmf2.8ml中,加入pd(oac)2(9.1mg、0.0403mmol)、1,2-双(二苯基膦基)乙烷(24.12mg、0.0604mmol)、et3n(168μl、1.21mmol)、苯甲醇(840μl、8.06mmol),在一氧化碳气氛下,回流24小时。反应后,减压蒸馏除去溶剂,溶解于乙酸乙酯中后,进行过滤,将滤液用5质量%碳酸氢钠水溶液洗涤。向有机层中加入硫酸钠并干燥后,减压蒸馏除去溶剂,将得到的残余物利用使用了氯仿和甲醇的flash色谱系统进行纯化,由此得到黄色油状物形态的化合物(a12)(113mg、0.166mmol、收率41.4%)。esi-ms(m+h)+:m/z767.50.found767.60.合成例l1(j):化合物(a13)将化合物(a12)(46mg、0.0600mmol)溶解于甲醇1ml中后,加入10质量%pd/c100mg,在氢气氛下,于室温搅拌2小时。过滤反应溶液后,减压蒸馏除去溶剂,由此得到黄色油状物形态的化合物(a13)(25.5mg、0.0377mmol、收率62.8%)。esi-ms(m+h)+:m/z677.45.found677.62.合成例l2:cdo3aet-fgk(化合物(a16)(与上述的化合物1-1相同)),cdo3aet-fgk-boc(化合物(a17)),cdo3aet-fgk(mal)(化合物(a18)(与上述的化合物1-2相同))的合成[化学式39](i)dic,hoat,dmf;tfa:tis:mq=95:2.5:2.5,2小时,(ii)(boc)2o二氧杂环己烷,饱和nahco3;(iii)饱和nahco3aq,(boc)2o,二氧杂环己烷,2小时(iv)n-甲氧基羰基马来酰亚胺,饱和nahco3aq合成例l2(i)及(ii):化合物(a16)将利用fmoc固相合成法将肽延伸而得到的树脂(a14)(22μmol)、1-羟基-7-氮杂苯并三唑(15.1mg、110μmol)、化合物(a13)(15mg、22μmol)溶解于dmf中,加入n,n'-二异丙基碳二亚胺(17.2μl、110μmol),于室温缓缓搅拌16小时。反应结束后使用dmf及ch2cl2洗涤树脂。将得到的树脂(a15)悬浮于以三氟乙酸:三异丙基硅烷:水=95:2.5:2.5作为成分的溶液中缓缓搅拌2小时。反应结束后,通过过滤除去树脂,减压蒸馏除去滤液,由此得到白色晶体。进而通过hplc,使用imtaktcadenza5cd-c18150×20mm,流动相中,a相使用0.1%tfa/milliq,b相使用0.1%tfa/mecn,通过线性梯度法,在0-35分钟内从a相95%、b相5%变化为a相70%、b相30%,在35-40分钟内从a相70%、b相30%变化为a相0%、b相100%,以流速5ml/分钟进行纯化,由此得到目标化合物(a16)(以下也称为“cdo3aet-fgk”)(5.7mg、6.78μmol、收率30.8%)。esi-ms(m-h)-:m/z839.4,found:839.3合成例l2(iii):化合物(a17)将化合物(a16)溶解于饱和碳酸氢钠水溶液100μl中,加入溶解有1.5当量的(boc)2o的二氧杂环己烷100μl,于室温剧烈搅拌两小时。通过减压蒸馏除去二氧杂环己烷后,利用氯仿洗涤水层。进而,对于水层,通过hplc,使用imtaktcadenza5cd-c18150×20mm,流动相中,a相使用0.1%tfa/milliq,b相使用0.1%tfa/mecn,将a相保持为100%直至2分钟为止,然后通过线性梯度法,在2-5分钟内从a相100%、b相0%变化为a相95%、b相5%,在5-40分钟内从a相95%、b相5%变化为a相70%、b相30%,在40-45分钟内从a相70%、b相30%变化为a相0%、b相100%,以流速5ml/分钟进行纯化,由此得到目标化合物(a17)(以下,也称为“cdo3aet-fgk-boc”)。esi-ms(m-h)-:m/z939.5,found:940.49合成例l2(iv):化合物(a18)cdo3aet-fgk(mal)将化合物(a16)(2.2mg、2.6μmol)溶解于饱和碳酸氢钠水溶液200μl中,在冰冷却下加入n-甲氧基羰基马来酰亚胺(0.8mg、5.2μmol),在冰冷却下搅拌两小时。反应结束后,使用10质量%柠檬酸水溶液调节为酸性。然后,通过hplc,使用imtaktcadenza5cd-c18150×20mm,流动相中,a相使用0.1%tfa/milliq,b相使用0.1%tfa/mecn,将a相保持为100%直至5分钟为止,然后通过线性梯度法,在5-35分钟内从a相100%、b相0%变化为a相55%、b相45%,在35-50分钟内从a相55%、b相45%变化为a相0%、b相100%,以流速5ml/分钟进行纯化,由此得到目标化合物(a18)(以下也称为“cdo3aet-fgk(mal)”)(0.8mg、0.870μmol、收率33.4%)。esi-ms(m-h)-:m/z919.42,found:919.45.[do3a-bn-scn-mvk(bzo)及do3a-bn-scn-met-oh,do3a-bn-scn-mvk(mal)的合成]合成例l3:化合物(b5)及化合物(b7)的合成[化学式40](a)溴乙酸叔丁酯,nahco3,乙腈,43.1%;(b)4-硝基苄溴,na2co3,乙腈,98.0%;(c)10%pd/c,甲醇,1nnaoh,87.2%;(d)10%苯甲醚/tfa,64.9%;(e)1m硫代羰基氯/氯仿,74.9%合成例l3(a):化合物(b1)的合成将大环多胺(523.6mg,3.04mmol)溶解于乙腈(25ml)中,加入nahco3(893.6mg,10.6mmol),在ar气氛下,冰冷却后,滴入溴乙酸叔丁酯(1.39ml,3.34mmol)。于室温搅拌48小时后,过滤反应液,将滤液减压蒸馏除去。将残余物利用使用了甲苯的重结晶进行纯化,由此得到白色晶体形态的化合物(b1)(672.4mg、收率43.1%)。1hnmr(cdcl3):δ1.44(27h,s,tbu),2.86-3.35(22h,overlapped,ch2).esi-ms(m+h)+:m/z515.3,found:515.3.合成例l3(b):化合物(b2)的合成将化合物(b1)(212.9mg,413.6μmol)溶解于乙腈(4.0ml)中,加入na2co3(87.7mg,827.4μmol),在ar气氛下,冰冷却后,滴入溶解于乙腈(1.0ml)中的4-硝基苄溴(134.1mg,620.7μmol)。于60℃搅拌18小时后,过滤反应液,减压蒸馏除去溶剂。将残余物利用以氯仿:甲醇=10:1作为洗脱溶剂的硅胶色谱进行纯化,由此得到黄色油状物质形态的化合物(b2)(263.4mg、收率98.0%)。1hnmr(cdcl3):δ1.36(27h,s,tbu),2.06-3.58(24h,overlapped,ch2),7.61-7.63(2h,d,aromatic),8.07-8.09(2h,d,aromatic).esi-ms(m+na)+:m/z672.4,found:672.3.合成例l3(c):化合物(b3)的合成将化合物(b2)(150mg,231μmol)溶解于甲醇(3.5ml)中后,加入1nnaoh(0.5ml)、10%pd/c(15.4mg)。氢气氛下,于室温搅拌1.5小时。过滤溶液后,从滤液中减压蒸馏除去甲醇,利用氯仿(5ml×3)萃取。向有机层中加入na2so4并干燥后,减压蒸馏除去溶剂,由此得到黄色油状物质形态的化合物(b3)(124.6mg、收率87.2%)。1hnmr(cdcl3):δ1.43(27h,s,tbu),2.85-3.35(24h,overlapped,ch2),6.59-6.61(2h,d,aromatic),6.93-6.95(2h,d,aromatic).esi-ms(m+h)+:m/z620.4,found:620.5.合成例l3(d):化合物(b4)的合成将化合物(b3)(124.6mg,201μmol)溶解于10%苯甲醚/三氟乙酸(tfa)(2ml)中,于室温搅拌4小时。减压蒸馏除去溶剂,加入乙醚(5ml),从而使其结晶化。过滤收集晶体,用乙醚洗涤后,减压干燥,由此得到红褐色晶体形态的化合物(b4)的tfa盐(118.4mg、收率64.9%)。1hnmr(d2o):δ2.50-3.50(24h,overlapped,ch2),6.78-6.81(2h,d,aromatic),7.36-7.38(2h,d,aromatic).合成例l3(e):化合物(b5)的合成将化合物(b4)(82.5mg,80.8μmol)溶解于milliq水(1ml)中,加入1m硫光气/氯仿(1ml)。于室温搅拌2小时后,用氯仿(5ml×4)洗涤。将水层冷冻干燥,由此得到淡黄色晶体形态的化合物(b5)(57.4mg、收率74.9%)。1hnmr(d2o):δ2.50-3.65(24h,overlapped,ch2),7.18-7.22(2h,d,aromatic),7.38-7.41(2h,d,aromatic).合成例l3(f):化合物(b6)的合成将化合物(b1)(100.0mg,194.4μmol)溶解于乙腈(2.0ml)中,加入na2co3(26.5mg,252.9μmol),在ar气氛下,冰冷却后,滴入溶解于乙腈(0.5ml)中的甲基-4-(溴甲基)苯甲酸酯(58.0mg,253.0μmol)。于60℃搅拌18小时后,过滤,减压蒸馏除去溶剂。将残余物利用以氯仿:甲醇=10:1作为洗脱溶剂的硅胶色谱进行纯化,由此得到黄色油状物质形态的化合物(b6)(106.1mg、收率82.6%)。1hnmr(cdcl3):δ1.46(27h,s,tbu),2.01-3.58(24h,overlapped,ch2),3.88(3h,s,och3),7.53-7.55(2h,d,aromatic),7.95-7.97(2h,d,aromatic).esi-ms(m+h)+:m/z663.4,found:663.4合成例l3(g):化合物(b7)的合成将化合物(b6)(106.1mg,160.0μmol)溶解于甲醇(1ml)中后,加入1nnaoh(1ml),于室温搅拌2小时。从溶液中减压蒸馏除去甲醇后,利用氯仿(5ml×3)萃取。向有机层中加入na2so4并干燥后,减压蒸馏除去溶剂,由此得到黄色油状物质形态的化合物(b7)(50.5mg、收率48.6%)。1hnmr(cdcl3):δ1.44(27h,s,tbu),2.13-3.58(24h,overlapped,ch2),7.31-7.33(2h,d,aromatic),8.09-8.11(2h,d,aromatic).esi-ms(m+na)+:m/z671.4,found:671.5合成例l4:do3a-bn-scn-mvk(bzo)(化合物(b13))及do3a-bn-scn-met-oh(化合物(b14))的合成[化学式41](a)n-羟基琥珀酰亚胺,dcc,二氯甲烷,74.7%;(b)fmoc-lys-oh,dipea,dmf,82.0%;(c)ⅰ.dipea,二氯甲烷;ⅱ.甲醇,dipea;ⅲ.20%哌啶/dmf;(d)fmoc固相延伸;(e)tfa:tis:h2o=95:2.5:2.5,69.0%;(f)p-scn-bn-do3a,0.16m硼酸盐缓冲液ph11.0,1nnaoh,53.3%;(g)甲硫氨酸,0.16m硼酸盐缓冲液ph11.0,1nnaoh,36.2%合成例l4(a):化合物(b8)的合成将苯甲酸(560mg,4.59mmol)和n-羟基琥珀酰亚胺(nhs,581mg,5.05mmol)溶解于ch2cl2(15ml)中,冰冷却后,滴入溶解于ch2cl2(8ml)中的n,n’-二环己基碳二亚胺(dcc,1.05g,5.05mmol)。于室温搅拌3小时后,过滤,减压蒸馏除去溶剂。将残余物溶解于乙酸乙酯(10ml)中,用饱和nahco3(10ml×3)洗涤。向有机层中加入mgso4并干燥后,减压蒸馏除去溶剂,由此得到白色晶体形态的化合物(b8)(749.7mg、收率74.7%)。1hnmr(cdcl3):δ2.89(4h,s,琥珀酰亚胺),7.47-7.51(2h,m,aromatic),7.64-7.68(1h,m,aromatic),8.11-8.13(2h,m,aromatic)合成例l4(b):化合物(b9)的合成将fmoc-lys-oh(86.7mg,100μmol)溶解于dmf(1.5ml)中,加入n,n-二异丙基乙胺(dipea,40μl,246μmol),在ar气氛下,冰冷却后,滴入溶解于dmf(0.5ml)中的化合物(b8)。于室温搅拌3小时后,减压蒸馏除去溶剂。将残余物溶解于乙酸乙酯(5ml)中,用10质量%柠檬酸(5ml×3)洗涤。向有机层中加入na2so4并干燥后,减压蒸馏除去溶剂,利用以氯仿:甲醇=5:1作为展开溶剂的tlc分离进行纯化,由此得到白色晶体形态的化合物(b9)(82.0mg、收率82.0%)。esi-ms(m+na)+:m/z473.2,found:473.2合成例l4(c):化合物(b10)的合成将cl-trt(2-cl)resin(62.5mg,100μmol、渡边化学工业株式会社)作为起始物质,使fmoc-lys(bzo)-oh(47.2mg,100μmol)及dipea(65μl,400μmol)于二氯甲烷(1.5ml)中反应1.5小时。加入甲醇(1.5ml)及dipea(65μl)停止反应。将树脂用dmf洗涤,然后用二氯甲烷洗涤。使树脂干燥后,通过测定在哌啶处理时生成的n-(9-芴基甲基)哌啶的a301处的吸光度来对fmoc-lys(bzo)-oh向树脂的导入量进行定量(0.96mmol/g)。加入20%哌啶/dmf(ml),于室温搅拌20分钟,由此制备化合物(b10)。采集一部分树脂进行kaiser试验,确认到nα-fmoc基的去保护。合成例l4(d):化合物(b11)的合成将化合物(b10)(22.9mg,22.0μmol)作为起始物质,利用fmoc固相合成法,将2.5等量的fmoc-met-oh(55.0μmol)、n,n’-二异丙基碳二亚胺(dic,8.5μl,55.0μmol)、1-羟基苯并三唑一水合物(hobt,8.43mg,55.0μmol)在dmf中于室温搅拌2小时。采集一部分树脂进行kaiser试验,以确认缩合反应的结束,然后,加入20%哌啶/dmf(2ml),于室温搅拌20分钟。采集一部分树脂进行kaiser试验,确认到nα-fmoc基的去保护。然后使用保护氨基酸boc-met-oh进行同样的操作,由此制备化合物(b11)。合成例l4(e):化合物(b12)的合成将化合物(b11)于tfa:三异丙基硅烷(tis):h2o=95:2.5:2.5(1ml)的混合液中于室温搅拌2小时。过滤除去树脂后,减压蒸馏除去滤液,向得到的残余物中加入乙醚以使其结晶化。过滤收集晶体,用乙醚洗涤后,减压干燥,由此得到白色晶体形态的化合物(b12)的tfa盐(9.7mg、收率69.0%)。esi-ms(m+h)+:m/z481.2,found:481.2.合成例l4(f):化合物(b13)的合成将化合物(b5)(1.0mg,1.05μmol)和化合物(b12)(1.58μmol)溶解于0.16m硼酸缓冲液ph11.0(100μl)中后,用1nnaoh调节为ph9.0。于室温搅拌2小时后,利用分离用rp-hplc进行纯化,得到白色晶体形态的化合物(b13)(以下也称为“do3a-bn-scn-mvk(bzo)”)的tfa盐(0.8mg、收率53.3%)。esi-ms(m+h)+:m/z974.4,found:974.4.合成例l4(g):化合物(b14)的合成将化合物(b5)(5.5mg,5.78μmol)和甲硫氨酸(1.29mg,8.67μmol)作为起始物质,进行与合成例l4(f)同样的操作,由此得到白色晶体形态的化合物(b14)(以下也称为“do3a-bn-scn-met-oh”)的tfa盐(2.3mg、收率36.2%)。esi-ms(m+h)+:m/z643.2,found:.643.2合成例l5:do3a-bn-scn-mvk(mal)(化合物(b21))的合成[化学式42](a)ⅰ.fmoc-lys(dde)-oh,dipea,二氯甲烷;ⅱ.甲醇,dipea;ⅲ.20%哌啶/dmf;(b)fmoc固相延伸;(c)2%hydrazine/dmf;(d)aceticacid:tfe:ch2cl2=3:1:6,93.9%;(e)nmcm,饱和nahco3,94.1%;(f)4mhcl/acoet,92.4%;(g)p-scn-bn-do3a,tea,dmf,33.2%合成例l5(a):化合物(b15)的合成将cl-trt(2-cl)resin(104.4mg,114.8μmol、渡边化学工业株式会社)作为起始物质,使fmoc-lys(dde)-oh(61.2mg,114.8μmol)及dipea(81μl,459.2μmol)于二氯甲烷(2ml)中反应1.5小时。加入甲醇及dipea停止反应。将树脂用dmf洗涤,然后用二氯甲烷洗涤。通过与合成例l4(c)同样的方法对fmoc-lys(bzo)-oh向树脂的导入量进行定量(0.769mmol/g)。加入20%哌啶/dmf(2ml),于室温搅拌20分钟,由此制备化合物(b15)。采集一部分树脂进行kaiser试验,确认到nα-fmoc基的去保护。合成例l5(b):化合物(b16)的合成将化合物(b15)(153.4mg,114.8μmol)作为起始物质,将保护氨基酸依次变更为fmoc-val-oh、boc-met-oh,进行与合成例l4(d)同样的操作,由此得到化合物(b16)。合成例l5(c):化合物(b17)的合成将化合物(b16)(117.9μmol)于2%肼/dmf(2ml)中于室温搅拌1小时后,采集一部分树脂进行kaiser试验,确认反应结束。将树脂用dmf洗涤,然后用二氯甲烷洗涤后,减压干燥,由此得到化合物(b17)。合成例l5(d):化合物(b18)的合成将化合物(b17)于乙酸:2,2,2-三氟乙醇(tfe):二氯甲烷=3:1:6(2ml)的混合液中于室温搅拌2小时。过滤除去树脂后,减压蒸馏除去滤液,向得到的残余物中加入乙醚以使其结晶化。过滤收集晶体,用乙醚洗涤后,减压干燥,由此得到白色晶体形态的化合物(b18)的乙酸盐(51.4mg、收率93.9%)。esi-ms(m+h)+:m/z477.2,found:477.2.合成例l5(e):化合物(b19)的合成将化合物(b18)(6.60mg,13.9μmol)通过冰冷却溶解于饱和nahco3(100μl)中,加入n-甲氧基羰基马来酰亚胺(nmcm,3.22mg,20.8μmol)。在冰冷却下搅拌2小时后,加入5质量%柠檬酸进行中和,利用氯仿(5ml×3)萃取。加入na2so4并干燥后,减压蒸馏除去溶剂,由此得到白色晶体形态的化合物(b19)(7.26mg、收率94.1%)。esi-ms(m+h)+:m/z557.2,found:557.2.合成例l5(f):化合物(b20)的合成将化合物(b19)(7.26mg,13.1μmol)溶解于4mhcl/乙酸乙酯(1ml)中,于室温搅拌1小时。减压蒸馏除去溶剂,与己烷共沸,由此得到白色晶体形态的化合物(b20)的盐酸盐(5.92mg、收率92.4%)。esi-ms(m+h)+:m/z457.2,found:457.2.合成例l5(g):化合物(b21)的合成将化合物(b20)的盐酸盐(3.38mg,4.74μmol)溶解于dmf(100μl)中,加入三乙胺(tea,6μl,43.3μmol)。加入化合物(b5)(3.0mg,3.16μmol),于室温搅拌2小时后,用h2o稀释为10倍,利用分离用rp-hplc进行纯化,由此得到白色晶体形态的化合物(b21)(以下也称为“do3a-bn-scn-mvk(mal)”)的tfa盐(1.5mg、收率33.2%)。esi-ms(m+h)+:m/z972.5,found:972.5.[do3a-bn-co-fgk、do3a-bn-co-fgk(boc)、do3a-bn-co-fgk(mal)、do3a-bn-co-phe-oh的合成]合成例l6:do3a-bn-co-fgk(化合物(b24)(与上述的化合物1-3相同))、do3a-bn-co-fgk(boc)(化合物(b25))、do3a-bn-co-fgk(mal)(化合物(b26)(与上述的化合物1-4相同))、do3a-bn-co-phe-oh(化合物(b28))的合成[化学式43](a)ⅰ.fmoc-lys(boc)-oh,dipea,二氯甲烷;ⅱ.甲醇,dipea;ⅲ.20%哌啶/dmf;(b)fmoc固相延伸;(c)ⅰ.p-cooh-bn-do3a,dic,hoat,dmf;ⅱ.tfa:tis:h2o=95:2.5:2.5,53.7%;(d)(boc)2o,饱和nahco3,26.9%;(e)nmcm,饱和nahco3,44.4%;(f)ⅰ.fmoc-phe-oh,dipea,二氯甲烷;ⅱ.甲醇,dipea;ⅲ.20%哌啶/dmf;(g)ⅰ.p-cooh-bn-do3a,dic,hoat,dmf;ⅱ.tfa:tis:h2o=95:2.5:2.5,59.1%合成例l6(a):化合物(b22)的合成将cl-trt(2-cl)resin(22.1mg,24.3μmol、渡边化学工业株式会社)作为起始物质,使fmoc-lys(boc)-oh(17.1mg,36.5μmol)及dipea(16.5μl,97.2μmol)于二氯甲烷(1ml)中反应1.5小时。加入甲醇及dipea停止反应。将树脂用dmf洗涤,然后用二氯甲烷洗涤。通过与合成例l4(c)同样的方法对fmoc-lys(boc)-oh向树脂的导入量进行定量(0.884mmol/g)。加入20%哌啶/dmf(2ml),于室温搅拌20分钟,由此制备化合物(b22)。采集一部分树脂进行kaiser试验,确认到nα-fmoc基的去保护。合成例l6(b):化合物(b23)的合成将化合物(b22)(12.1μmol)作为起始物质,将保护氨基酸依次变更为fmoc-gly-oh、fmoc-phe-oh,进行与合成例l4(d)同样的操作,由此得到化合物(b23)。合成例l6(c):化合物(b24)的合成对于化合物(b23)(12.1μmol),将化合物(b7)(15.7mg,24.2μmol)、dic(3.7μl,24.2μmol)、hoat(3.29mg,24.2μmol)于dmf中,于室温搅拌一夜。采集一部分树脂进行kaiser试验,以确认缩合反应的结束,然后,于tfa:tis:h2o=95:2.5:2.5(1ml)的混合液中于室温搅拌2小时。过滤除去树脂后,减压蒸馏除去滤液,向得到的残余物中加入乙醚以进行结晶化。过滤收集晶体,用乙醚洗涤后,减压干燥,由此得到白色晶体形态的化合物(b24)(以下也称为“do3a-bn-co-fgk”)的tfa盐(9.0mg、收率53.7%)。esi-ms(m+h)+:m/z813.4,found:813.4.合成例l6(d):化合物(b25)的合成将化合物(b24)(2.17μmol)溶解于饱和nahco3(100μl)中后,加入溶解于二氧杂环己烷(100μl)中的(boc)2o(7.1mg,3.26μmol),于室温剧烈搅拌2小时。减压蒸馏除去二氧杂环己烷后,用氯仿(3ml×3)洗涤。将水层利用分离用rp-hplc进行纯化,由此得到白色晶体形态的化合物(b25)(以下也称为“do3a-bn-co-fgk(boc)”)的tfa盐(0.8mg、收率26.9%)。esi-ms(m+h)+:m/z913.5,found:913.5.合成例l6(e):化合物(b26)的合成将化合物(b24)(2.17μmol)溶解于饱和nahco3(100μl)中后,冰冷却并加入nmcm(0.5mg,3.26μmol)。在冰冷却下搅拌2小时后,利用分离用rp-hplc进行纯化,由此得到白色晶体形态的化合物(b26)(以下也称为“do3a-bn-co-fgk(mal)”)的tfa盐(1.3mg、收率44.4%)。esi-ms(m+h)+:m/z893.4,found:893.4.合成例l6(f):化合物(b27)的合成将cl-trt(2-cl)resin(5mg,5.50μmol、渡边化学工业株式会社)作为起始物质,使fmoc-phe-oh(6.01μmol)及dipea(3.73μl,21.9μmol)于二氯甲烷(0.5ml)中反应1.5小时。加入甲醇及dipea停止反应。将树脂用dmf洗涤,然后用二氯甲烷洗涤。通过与合成例l4(c)同样的方法对fmoc-a.a.-oh向树脂的导入量进行定量(0.917mmol/g)。加入20%哌啶/dmf(2ml),于室温搅拌20分钟,由此制备化合物(b27)。采集一部分树脂进行kaiser试验,确认到nα-fmoc基的去保护。合成例l6(g):化合物(b28)的合成将化合物(b27)(5.5μmol)作为起始物质,通过与合成例l6(c)同样的操作,得到白色晶体形态的化合物(b28)(以下也称为“do3a-bn-co-phe-oh”)的tfa盐(4.5mg、收率59.1%)。esi-ms(m+h)+:m/z628.3,found:628.3.[cdota-bn-co-fgk、cdota-bn-co-fgk(boc)、cdota-bn-co-fgk(mal)的合成]合成例l7:cdota-bn-co-fgk(化合物(b29))、cdota-bn-co-fgk(boc)(化合物(b30))、cdota-bn-co-fgk(mal)(化合物(b31))的合成[化学式44](a)ⅰ.p-cooh-bn-dota(tbu)4,dic,hoat,dmf;ⅱ.tfa:tis:h2o=95:2.5:2.5,1.3%;(d)(boc)2o,饱和nahco3,%;(e)nmcm,饱和nahco3,61.4%合成例l7(a):化合物(b29)的合成将p-cooh-bn-dota(tbu)4(52.4μmol)作为起始物质,通过与合成例l6(c)同样的操作,得到白色晶体形态的化合物(b29)(以下也称为“cdota-bn-co-fgk”)的tfa盐(2.4mg、收率1.3%)。esi-ms([m+k]-h)-:m/z907.36,found:907.31.合成例l7(b):化合物(b30)的合成将化合物(b29)(5.75μmol)作为起始物质,通过与合成例l6(d)同样的操作,得到白色晶体形态的化合物(b30)(以下也称为“cdota-bn-co-fgk(boc)”)的tfa盐(0.4mg、收率71.8%)。esi-ms([m+k]-h)-:m/z1007.41,found:1007.31.合成例l7(c):化合物(b31)的合成将化合物(b29)(0.694μmol)作为起始物质,通过与合成例l6(e)同样的操作,得到白色晶体形态的化合物(b31)(以下也称为“cdota-bn-co-fgk(mal)”)的tfa盐(0.6mg、收率61.4%)。esi-ms(m-h)-:m/z949.39,found:949.43.[cdo3aibu-fgk(boc),cdo3aibu-fgk(mal),cdo3aibu-phe的合成]合成例l8:化合物(a23)的合成[化学式45](a)异丁醛,三乙酰氧基硼氢化钠,thf,42.4%;(b)1mbh3-thf,thf,74.7%;(c)溴乙酸叔丁酯,na2co3,乙腈,58.8%;(d)pd(oac)2,1,2-双(二苯基膦基)乙烷,et3n,co,bnoh,dmf,25.5%;(j)10%pd/c,meoh,48.8%.合成例l8(a):化合物(a19)的合成将化合物(a8)(913mg、1.96mmol)溶解于thf(20ml)中,加入异丁醛(357μl、3.91mmol)、三乙酰氧基硼氢化钠(498mg、2.35mmol),在ar气氛下,于室温搅拌一夜。加入异丁醛(179μl、1.96mmol)、三乙酰氧基硼氢化钠(249mg、1.18mmol)后,进而于室温搅拌2小时。将反应液冰冷却,加入水后,减压蒸馏除去thf,从得到的水溶液中用氯仿萃取三次。向有机层中加入硫酸钠并干燥后,减压蒸馏除去溶剂,将得到的残余物利用使用了氯仿和甲醇的flash色谱系统进行生成,由此得到白色晶体形态的化合物(a19)(403mg、829μmol,42.4%)。1hnmr(cdcl3):δ0.92-0.95(6h,m,ch3),1.68-1.71(1h,m,ch),2.40-2.44(2h,m,ch2),2.82-3.30(8h,overlapped,ch2),3.61-3.74(2h,m,ch2),4.58-4.62(1h,m,ch),6.48(1h,s,nh),6.66(1h,s,nh),6.96-6.98(2h,d,ch2),7.56-7.58(2h,d,ch2),8.00(1h,s,nh).esi-ms(m+h)+:m/z487.12,found:487.18.合成例l8(b):化合物(a20)的合成将化合物(a19)(403mg、829μmol)悬浮于thf2ml中,将溶液冰冷却后,在氩气气氛下,缓缓加入0.95m的硼烷-thf络合物/thf溶液13ml,搅拌1小时后,回流22小时。将溶液冰冷却,加入甲醇13ml后,搅拌1小时,减压蒸馏除去溶剂。然后,向残余物中再次加入甲醇13ml,减压蒸馏除去溶剂。向残余物中加入浓盐酸13ml,于室温搅拌24小时后,回流1小时。将溶液冰冷却,加入12.5n氢氧化钠水溶液使其成为碱性后,用氯仿萃取。向有机层中加入硫酸钠并干燥后,减压蒸馏除去溶剂,将得到的残余物利用以氯仿:甲醇:25质量%氨水(10:1:0.1)作为洗脱溶剂的flash色谱系统进行纯化,由此得到黄色油状物形态的化合物(a20)(275mg、619μmol、收率74.7%)。1hnmr(cdcl3):δ0.89-0.92(6h,m,ch3),1.80-1.84(1h,m,ch),2.06-2.89(19h,overlapped,ch,ch2),6.92-6.94(2h,d,ch2),7.58-7.60(2h,d,ch2).合成例l8(c):化合物(a21)的合成将化合物(a20)(275mg、619μmol)悬浮于乙腈2ml中,加入碳酸钠(428mg、3.10mmol)。将溶液冰冷却,在氩气气氛下,滴入溴乙酸叔丁酯(272μl、1.86mmol)。滴入完成后,于室温搅拌24小时,过滤悬浮液后,从滤液中减压蒸馏除去溶剂。将残余物利用使用了氯仿和甲醇的flash色谱系统进行生成,由此得到黄色油状物形态的化合物(a21)(286mg、364μmol、58.8%)。1hnmr(cdcl3):δ0.86-0.89(6h,m,ch3),1.41-1.50(27h,m,tbu),1.86-3.90(26h,overlapped,ch,ch2),6.99-7.01(2h,d,ch2),7.59-7.61(2h,d,ch2).esi-ms(m+h)+:m/z787.39,found:787.45.合成例l8(d):化合物(a22)的合成将化合物(a21)(286mg、364μmol)悬浮于dmf3.0ml中,加入pd(oac)2(16.3mg、0.0728mmol)、1,2-双(二苯基膦基)乙烷(58.0mg、0.146mmol)、et3n(156μl、1.12mmol)、苯甲醇(753μl、7.28mmol),在一氧化碳气氛下,回流一夜。反应后,减压蒸馏除去溶剂,将得到的残余物利用使用了氯仿和甲醇的flash色谱系统进行纯化,由此得到黄色油状物形态的化合物(a22)(73.8mg、92.8μmol、收率25.5%)。esi-ms(m+h)+:m/z795.53,found:795.40.合成例l8(e):化合物(a23)的合成将化合物(a22)(73.8mg、92.8μmol)溶解于甲醇1.5ml中后,加入10质量%pd/c150mg,在氢气氛下,于室温搅拌5小时。过滤反应溶液后,减压蒸馏除去溶剂,由此得到白色晶体形态的化合物(a23)(31.9mg、45.3μmol、收率48.8%)。esi-ms(m+h)+:m/z705.48,found:705.40.合成例l9:化合物(a28)的合成[化学式46](a)2,3,5,6-四氟苯酚,edc,et3n,dmf,68.9%;(b)phe-gly-lys(boc)-trt(2-cl)-resin,diea,dmf;(c)acoh:2,2,2-三氟乙醇:ch2cl2=3:1:6,2.1%;(d)tfa:tis:水=95:2.5:2.5,6.4%;(e)n-甲氧基羰基马来酰亚胺,饱和nahco3,45.5%:(f)i)h-phe-otbu·hcl,comu,diea,dmf,ii)10%苯甲醚/tfa,2.3%.合成例l9(a):化合物(a24)的合成将化合物(a23)(23.7mg、33.6μmol)溶解于dmf0.9ml中后,加入2,3,5,6-四氟苯酚(8.4mg、50.6μmol)、三乙胺(9.3μl、66.9μmol)。将溶液冰冷却,加入edc(9.6mg、50.6μmol)后,恢复至室温,搅拌4小时。减压蒸馏除去溶剂后,将残余物溶解于中乙酸乙酯,用饱和氯化铵水溶液洗涤三次。向有机层中加入硫酸钠并干燥后,减压蒸馏除去溶剂,由此得到黄色油状物形态的化合物(a24)(18.3mg、23.2μmol、收率68.9%)。合成例l9(b)及(c):化合物(a26)的合成将利用fmoc固相合成法将肽延伸而得到的树脂(64.0mg、57.6μmol)、n,n-二异丙基乙胺(23.6μl、135μmol)、化合物(a24)(18.3mg、23.2μmol)溶解于dmf中,于室温缓缓搅拌16小时。反应结束后使用dmf及ch2cl2洗涤树脂。将得到的树脂(a25)悬浮于以乙酸:2,2,2-三氟乙醇:二氯甲烷=3:1:6作为成分的溶液中缓缓搅拌2小时。反应结束后,通过过滤除去树脂,减压蒸馏除去滤液,然后与甲苯共沸三次。将残余物利用hplc使用imtaktcadenza5cd-c18150×20mm,流动相中,a相使用0.1%tfa/milliq,b相使用0.1%tfa/mecn,通过线性梯度法,在0-35分钟内从a相95%、b相5%变化为a相50%、b相50%,在35-45分钟内从a相50%、b相50%变化为a相0%、b相100%,以流速5ml/分钟进行纯化(保留时间为34.7分钟),由此得到目标化合物(a26)(以下也称为“cdo3aibu-fgk(boc)”)(1.0mg、0.70μmol、收率2.1%)。esi-ms(m+h)+:m/z969.53,found:969.51.合成例l9(d):化合物(a27)的合成将使用利用fmoc固相合成法将肽延伸而得到的树脂(48.3mg、43.4μmol)、n,n-二异丙基乙胺(17.8μl、102μmol)、化合物(a24)(18.3mg、17.5μmol)制备的、通过与上述同样的方法制备的树脂(a25)悬浮于以三氟乙酸:三异丙基硅烷:水=95:2.5:2.5作为成分的溶液中,缓缓搅拌2小时。反应结束后,通过过滤除去树脂,减压蒸馏除去滤液,然后与甲苯共沸三次。将残余物溶解于水中,用乙醚洗涤三次,将得到的水溶液冷冻干燥,将得到的白色晶体通过hplc,使用imtaktcadenza5cd-c18150×20mm,流动相中,a相使用0.1%tfa/milliq,b相使用0.1%tfa/mecn,通过线性梯度法,在0-35分钟内从a相95%、b相5%变化为a相50%、b相50%,在35-45分钟内从a相50%、b相50%变化为a相0%、b相100%,以流速5ml/分钟进行纯化(保留时间为24.8分钟),由此得到目标化合物(a27)(以下也称为“cdo3aibu-fgk”)(1.8mg、1.25μmol、收率6.4%)。esi-ms(m+h)+:m/z869.48,found:869.38.合成例l9(e):化合物(a28)的合成例将化合物(a27)(1.8mg、2.6μmol)溶解于饱和碳酸氢钠水溶液150μl中,在冰冷却下加入n-甲氧基羰基马来酰亚胺(1.0mg、6.5μmol),在冰冷却下搅拌两小时。反应结束后,用5质量%柠檬酸水溶液调节为酸性。然后通过hplc,使用imtaktcadenza5cd-c18150×20mm,流动相中,a相使用0.1%tfa/milliq,b相使用0.1%tfa/mecn,将a相保持为100%直至5分钟后,通过线性梯度法,在5-35分钟内从a相100%、b相0%变化为a相40%、b相60%,在35-50分钟内从a相40%、b相60%变化为a相0%、b相100%,以流速5ml/分钟进行纯化(保留时间为30.7分钟),由此得到目标化合物(a28)(以下也称为“cdo3aibu-fgk(mal)”)(0.8mg、0.57μmol、收率45.5%)。esi-ms(m+h)+:m/z949.47,found:949.36.合成例l9(f):化合物(a29)的合成将化合物(a23)(2.7mg、3.83μmol)溶解于dmf(0.3ml)中,添加h-phe-otbu·hcl(1.5mg、5.75μmol)、n,n-二异丙基乙胺(2.9μl、17.2μmol)、(1-氰基-2-乙氧基-2-氧代乙叉基氨基氧基)二甲基氨基-吗啉基-六氟磷酸碳鎓盐(4.9mg、11.5μmol),于室温搅拌一夜。减压蒸馏除去溶剂后,将残余物再次溶解于乙酸乙酯中,用5质量%碳酸氢钠水溶液洗涤三次,用5质量%柠檬酸水溶液洗涤三次。向有机层中加入硫酸钠并干燥后,减压蒸馏除去溶剂,向由此得到的残余物中加入10质量%苯甲醚/tfa溶液(2.0ml),于室温搅拌2小时。减压蒸馏除去溶剂后,与甲苯共沸三次。将残余物溶解于水中,用乙醚洗涤三次,从得到的水溶液中减压蒸馏除去溶剂,将得到的白色晶体通过hplc,使用imtaktcadenza5cd-c18150×20mm,流动相中,a相使用0.1%tfa/milliq,b相使用0.1%tfa/mecn,通过线性梯度法,在0-30分钟内从a相100%、b相0%变化为a相40%、b相60%,在30-35分钟内从a相40%、b相60%变化为a相0%、b相100%,以流速5ml/分钟进行纯化(保留时间为25.2分钟),由此得到目标化合物(a29)(以下也称为“cdo3aibu-phe”)(0.1mg、8.8μmol、收率2.3%)。esi-ms(m+h)+:m/z684.36ound:684.27[二官能性螯合剂结合fab的制备]合成例f1:fab(来源于兔血清igg)及it-fab(来源于兔血清igg)的制备(fab(来源于兔血清igg)的制备)将兔血清igg(9mg)溶解于含有10mmna2edta及20mm半胱氨酸的20mm磷酸缓冲液(ph7.0)1.5ml中,加入固定化木瓜蛋白酶50%浆液(thermoscientific,yokohama,japan)500μl,于37℃孵育42小时。反应结束后,加入10mm的tris盐酸缓冲液(ph7.5)2ml,用0.45μm的过滤器过滤,回收滤液。使用10kda超滤膜将滤液替换为20mm磷酸缓冲液(ph7.0),浓缩至1ml。此后,使用蛋白a柱进行纯化以获得fab。通过将0.1m磷酸缓冲液(ph6.8)作为洗脱溶剂并以流速1.0ml/分钟洗脱的se-hplc来确认获得的fab的生成,通过测定a280来算出浓度。(it-fab(来源于兔血清igg)的制备)使用含有充分脱气的2mmedta的0.16m硼酸缓冲液(ph8.0)以制备fab溶液(100μl,5mg/ml),一边搅拌一边按每次1μl加入溶解于相同的缓冲液中的2-亚氨基硫杂环戊烷(2-it)(5μl,2.88mg/ml),于37℃静静搅拌30分钟。反应后,使用旋转柱法[analyticalbiochemistry,1984,142,68-78]除去反应溶液中过量的2-it,得到it-fab(来源于兔血清igg)溶液,所述旋转柱法使用通过含有充分脱气的2mmedta的0.im磷酸缓冲液(ph6.0)平衡化后的sephadexg-50fine。使用2,2’-联吡啶二硫化物测定导入至每一分子fab的巯基的数量[archivesofbiochemistryandbiophysics,1967,119,41-49]。合成例f2:fab(来源于抗c-kitigg)及it-fab(来源于抗c-kitigg)的制备除了使兔血清igg(9mg)为抗c-kitigg(9mg)以外,通过与合成例f1同样的方法,制备fab(来源于抗c-kitigg)及it-fab(来源于抗c-kitigg)。[结合有fab的配体的制备]合成例:cdo3aet-fgk-fab(来源于兔血清igg)、cdo3aet-fgk-fab(来源于抗c-kitigg)、do3a-eda-fab(来源于兔血清igg)、cdota-bn-co-fgk-fab(来源于抗c-kitigg)、do3a-bn-scn-mvk-fab(来源于兔血清igg)、do3a-bn-co-fgk-fab(来源于兔血清igg)、cdo3aibu-fgk-fab(来源于兔血清igg)及cdo3aibu-fgk-fab(来源于抗c-kitigg)的制备向it-fab(来源于兔血清igg)溶液(100μl)中按每次1μl加入溶解于h2o中的cdo3aet-fgk(mal)(5μl,相对于巯基而言为20等量),于37℃反应2小时。然后,使用0.1m磷酸缓冲液(ph6.0)制备碘乙酰胺溶液,将其以相对于残留的巯基而言为500等量添加后,于37℃反应1小时,将未反应的巯基进行烷基化。然后,使用通过0.25m乙酸缓冲液(ph5.5)平衡化后的sephadexg-50fine的旋转柱法进行纯化,得到cdo3aet-fgk-fab(来源于兔血清igg)溶液。导入至每一分子fab的来源于cdo3aet-fgk(mal)的单元数通过将在加入碘乙酰胺之前使用dps测得的[archivesofbiochemistryandbiophysics,1967,119,41-49]硫醇数减去先前求得的硫醇数来求得。将cdo3aet-fgk(mal)分别变更为do3a-eda(mal)、do3a-bn-scn-mvk(mal)、do3a-bn-co-fgk(mal)、及cdo3aibu-fgk(mal),除此以外,通过与上述同样的方法,得到do3a-eda-fab(来源于兔血清igg)溶液、do3a-bn-scn-mvk-fab(来源于兔血清igg)溶液、do3a-bn-co-fgk-fab(来源于兔血清igg)溶液、及cdo3aibu-fgk-fab(来源于兔血清igg)溶液。将it-fab(来源于兔血清igg)溶液变更为it-fab(来源于抗c-kitigg)溶液,除此以外,通过与上述同样的方法,得到cdo3aet-fgk-fab(来源于抗c-kitigg)溶液。将it-fab(来源于兔血清igg)溶液变更为it-fab(来源于抗c-kitigg)溶液,将cdo3aet-fgk(mal)变更为cdota-bn-co-fgk(mal),除此以外,通过与上述同样的方法,得到cdota-bn-co-fgk-fab(来源于抗c-kitigg)溶液。将it-fab(来源于兔血清igg)溶液变更为it-fab(来源于抗c-kitigg)溶液,将cdo3aet-fgk(mal)变更为cdo3aibu-fgk(mal),除此以外,通过与上述同样的方法,得到cdo3aibu-fgk-fab(来源于抗c-kitigg)溶液。合成例:dota-bn-scn-fab(来源于抗c-kitigg)的制备使用0.16m硼酸缓冲液(ph8.5)制备fab溶液(100μl,5.0mg/ml),加入溶解于相同缓冲液中的dota-bn-scn(macrocyclics,usa)(14μl,10mg/ml),于4℃静置一夜。反应后,通过使用充分脱气的0.25m乙酸缓冲液(ph5.5)平衡化后的sephadexg-50fine的旋转柱法除去过量的dota-bn-scn,得到dota-bn-scn-fab(来源于抗c-kitigg)。需要说明的是,上述的cdo3aet-fgk-fab、do3a-bn-co-fgk-fab、cdota-bn-co-fgk-fab、cdo3aibu-fgk-fab的化学结构如上所述。另外,do3a-eda-fab、do3a-bn-scn-mvk-fab、及dota-bn-scn-fab的化学结构如下所述。[化学式47][化学式48][化学式49][金属络合物的制备]合成例:111in-cdo3aet-fgk-fab(来源于兔血清igg)、111in-cdo3aet-fgk-fab(来源于抗c-kitigg)、111in-do3a-eda-fab(来源于兔血清igg)、111in-cdota-bn-co-fgk-fab(来源于抗c-kitigg)、111in-do3a-bn-scn-mvk-fab(来源于兔血清igg)、111in-do3a-bn-co-fgk-fab(来源于兔血清igg)、111in-cdo3aibu-fgk-fab(来源于兔血清igg)、111in-cdo3aibu-fgk-fab(来源于抗c-kitigg)、111in-dota-bn-scn-fab(来源于抗c-kitigg)、111in-cdo3aet-fgk(boc)、111in-cdota-bn-co-fgk(boc)、111in-do3a-bn-scn-mvk(bzo)、及111in-do3a-bn-co-fgk(boc)的制备将111incl3(45μl)与1m乙酸缓冲液(ph5.5,5μl)混和,于室温静置5分钟。混合cdo3aet-fgk-fab(来源于兔血清igg)溶液(30μl)后,于40℃孵育90分钟。以最终浓度成为10mm的方式加入dtpa后,于室温静置18小时。通过使用0.1md-pbs(ph7.4)平衡化后的sephadexg-50fine的旋转柱法进行纯化,由此制备111in-cdo3aet-fgk-fab。将cdo3aet-fgk-fab(来源于兔血清igg)溶液分别变更为cdo3aet-fgk-fab(来源于抗c-kitigg)溶液、do3a-eda-fab(来源于兔血清igg)溶液、cdota-bn-co-fgk-fab(来源于抗c-kitigg)溶液、do3a-bn-scn-mvk-fab溶液(来源于兔血清igg)、do3a-bn-co-fgk-fab(来源于兔血清igg)溶液、cdo3aibu-fgk-fab(来源于兔血清igg)溶液、dota-bn-scn-fab(来源于抗c-kitigg)溶液、cdo3aet-fgk(boc)溶液、cdota-bn-co-fgk(boc)溶液、do3a-bn-scn-mvk(bzo)溶液、及do3a-bn-co-fgk(boc)溶液,除此以外,通过与上述同样的方法,制备111in-cdo3aet-fgk-fab(来源于抗c-kitigg)、111in-do3a-eda-fab(来源于兔血清igg)、111in-cdota-bn-co-fgk-fab(来源于抗c-kitigg)、111in-do3a-bn-scn-mvk-fab(来源于兔血清igg)、111in-do3a-bn-co-fgk-fab(来源于兔血清igg)、111in-cdo3aibu-fgk-fab(来源于兔血清igg)、111in-dota-bn-scn-fab(来源于抗c-kitigg)、111in-cdo3aet-fgk(boc)、111in-cdota-bn-co-fgk(boc)、111in-do3a-bn-scn-mvk(bzo)、及111in-do3a-bn-co-fgk(boc)、。[特性的研究]〔与bbmvs的孵育试验〕(肾刷状缘膜囊泡)按照hori等人的方法,从wistar系雄性大鼠(200-250g)的肾脏制备肾刷状缘膜囊泡(bbmvs)[biochemicalpharmacology45:1763-1768,1993]。所有操作均在冰上进行。向皮质中加入按重量计为4-5倍的300mm甘露醇、及含有5mmegta的12mmtris-盐酸缓冲液(ph7.1),并用polytron均化器(pt-3100kinematicagmghlittau,switzerland)均质化2分钟,并用相同的缓冲液进行稀释以制成10%匀浆。然后,在用蒸馏水稀释为2倍后,加入调节为1.0m的mgcl2水溶液以使最终浓度为10mm,放置15分钟。然后,将匀浆以1,900g离心,将上清液再次以24,000g离心30分钟。将沉淀再次悬浮于相当于皮质重量的20倍的150mm甘露醇、及含有2.5mmegta的6mmtris-盐酸缓冲液(ph7.1)中,用teflon(注册商标)均化器(1,000rpm,10次(strokes))进行均质化。然后,加入1.0m的mgcl2水溶液以使最终浓度为10mm,将悬浮液放置15分钟,然后,将匀浆以1,900g离心,将上清液再次以24,000g离心30分钟。将得到的沉淀悬浮于相当于皮质重量的10倍的0.1m磷酸缓冲液(ph7.0)中,再次利用teflon(注册商标)均化器(1,000rpm,10strokes)进行均质化。然后,将匀浆以24,000g离心30分钟,以沉淀形式得到bbmvs。然后,将bbmvs的沉淀再次悬浮于0.1m磷酸缓冲液(ph7.0)中,使其在0.4×19mm的针中通过十次,由此使囊泡的大小保持恒定。在孵育实验中,以蛋白质浓度成为10mg/ml的方式进行稀释并使用。关于制得的bbmvs,通过使用对硝基苯基-β-d-半乳糖吡喃糖苷来测定作为溶酶体标记酶的β-半乳糖苷酶活性,由此来评价溶酶体酶的混入[plantphysiology55:94-98,1975]。另外,按照glossmann等人[febsletters19:340-344,1972]、kramers等人[europeanjournalofbiochemistry99:345-351,1979]的方法,使用l-γ-谷氨酰-对硝基苯胺、l-亮氨酸-对硝基苯胺测定γ-谷氨酰转移酶、氨肽酶的活性。(孵育试验)bbmvs与111in标记小分子模型底物的孵育实验通过以下的方法进行。将以蛋白质浓度成为10mg/ml的方式制备的bbmvs(10μl)于37℃预孵育10分钟。然后,通过反相hplc除去过量的配体,加入溶解于pbs中的111in-cdo3aet-fgk(boc)溶液(10μl),于37℃孵育两小时。向bbmvs溶液中加入乙醇以使得乙醇浓度成为60%,以5000rpm离心10分钟。回收上清液后,向沉淀加入60%乙醇,再次使用同样的方法离心后,回收上清液。将得到的上清液通过hplc,使用imtaktunisonus-c18150×4.6mm,流动相中,a相使用0.1%tfa/milliq,b相使用0.1%tfa/mecn,通过线性梯度法,在0-30分钟内从a相100%、b相0%变化为a相55%、b相45%,以流速1ml/分钟进行分析。图1示出了111in-cdo3aet-fgk(boc)与bbmvs的孵育的实验结果。将111in-cdo3aet-fgk(boc)溶液替换为111in-cdota-bn-co-fgk(boc)溶液、111in-do3a-bn-co-fgk(boc)溶液、111in-do3a-bn-scn-mvk(bzo)溶液、111in-cdo3aibu-fgk(boc)溶液,进行同样的孵育实验、以及不使用bbmvs进行对照实验。图2示出了111in-cdota-bn-co-fgk(boc)溶液与bbmvs的孵育的实验结果。图3示出了111in-do3a-bn-scn-mvk(bzo)与bbmvs的孵育的实验结果。图4示出了111in-do3a-bn-co-fgk(boc)与bbmvs的孵育的实验结果。图5示出了111in-cdo3aibu-fgk(boc)与bbmvs的孵育的实验结果。由上述的实验结果可以理解,作为连接基团,与本发明的化合物同样地具有苄基酰胺结构的111in-cdo3aet-fgk(boc)、111in-cdota-bn-co-fgk(boc)、111in-do3a-bn-co-fgk(boc)、111in-cdo3aibu-fgk(boc)中,通过bbmvs的孵育从而使螯合配体部位游离。与此相对,具有硫代脲结构作为连接基团的化合物111in-do3a-bn-scn-mvk(bzo)中,未观测到游离物。〔金属络合物的小鼠血浆中稳定性的研究〕向小鼠血浆(90μl)中加入溶解于pbs中的111in-cdo3aet-fgk-fab(来源于兔血清igg)(10μl),于37℃孵育。在1、3、6、24小时后采集一部分溶液,利用以甲醇:10质量%乙酸铵水溶液=3:2作为展开溶剂的rp-tlc进行分析,由此算出原型(111in-cdo3aet-fgk-fab)的放射活性的比例,示于表1。[表1]表1原型百分比(%)1h95.5±0.313h95.5±0.896h95.6±0.6324h94.8±0.58将111in-cdo3aet-fgk-fab(来源于兔血清igg)变更为111in-cdo3aet-fgk-fab(来源于抗c-kitigg),除此以外,通过与上述同样的方法,实施小鼠血浆中稳定性试验。孵育2小时后,原型(111in-cdo3aet-fgk-fab)的放射活性的比例为95.2±0.3%。将111in-cdo3aet-fgk-fab(来源于兔血清igg)变更为111in-cdota-bn-co-fgk-fab(来源于抗c-kitigg),除此以外,通过与上述同样的方法,实施小鼠血浆中稳定性试验。孵育2小时后,原型(111in-cdota-bn-co-fgk-fab)的放射活性的比例为95.2±0.3%。将111in-cdo3aet-fgk-fab(来源于兔血清igg)变更为111in-cdo3aibu-fgk-fab(来源于兔血清igg),除此以外,通过与上述同样的方法,实施小鼠血浆中稳定性试验。表2示出了实验结果。[表2]表2原型百分比(%)1h92.0±0.1%6h93.6±0.3%24h91.4±1.6%〔金属络合物的小鼠药物动力学的研究〕将实施例及比较例中制备的金属络合物使用d-pbs(-)(ph7.4)稀释。将未修饰fab浓度调节为5μg/100μl的111in-cdo3aet-fgk-fab(来源于兔血清igg)溶液(0.3μci/100μl/只)通过对6周龄的ddy系雄性小鼠进行尾静脉注射来施予。在施予后10、30分钟、1、3、6、24小时后处死各组3只小鼠,采集重要器官,测定其重量后,通过自动井伽马系统测定放射活性。另外,分别采集到6、24小时后为止的粪便和尿液,测定放射活性。通过同样的方法,使用111in-cdo3aet-fgk-fab(来源于抗c-kitigg)溶液,通过6周龄的ddy系雄性小鼠尾静脉注射,针对重要器官、粪便和尿液测定放射活性。作为对照化合物,使用了通过同样的方法制备的111in-do3a-eda-fab(来源于兔血清igg)。表3示出了利用111in-cdo3aet-fgk-fab(来源于兔血清igg)的小鼠体内的放射活性的测定结果。表4示出了利用111in-cdo3aet-fgk-fab(来源于抗c-kitigg)的小鼠体内的放射活性的测定结果。表5示出了利用111in-do3a-eda-fab(来源于兔血清igg)的小鼠体内的放射活性的测定结果。通过与上述同样的方法,使用111in-cdota-bn-co-fgk-fab(来源于抗c-kitigg)溶液、111in-do3a-bn-scn-mvk-fab(来源于兔血清igg)溶液、111in-do3a-bn-co-fgk-fab(来源于兔血清igg)溶液、111in-cdo3aibu-fgk-fab(来源于兔血清igg)溶液、111in-cdo3aibu-fgk-fab(来源于抗c-kitigg)溶液、111in-dota-bn-scn-fab(来源于抗c-kitigg)溶液,通过6周龄的ddy系雄性小鼠尾静脉注射,针对重要器官、粪便和尿液测定放射活性。表6示出了利用111in-cdota-bn-co-fgk-fab(来源于抗c-kitigg)的小鼠体内的放射活性的测定结果。表7示出了利用111in-do3a-bn-scn-mvk-fab(来源于兔血清igg)的小鼠体内的放射活性的测定结果。表8示出了利用111in-do3a-bn-co-fgk-fab(来源于兔血清igg)的小鼠体内的放射活性的测定结果。表9示出了利用111in-cdo3aibu-fgk-fab(来源于兔血清igg)的小鼠体内的放射活性的测定结果。表10示出了利用111in-cdo3aibu-fgk-fab(来源于抗c-kitigg)的小鼠体内的放射活性的测定结果。表11示出了利用111in-dota-bn-scn-fab(来源于抗c-kitigg)的小鼠体内的放射活性的测定结果。图6a~c示出了111in-cdo3aet-fgk-fab与111in-do3a-eda-fab的结果的比较。由上述本发明的化合物即111in-cdo3aet-fgk-fab与比较化合物即111in-do3a-eda-fab的结果可知,111in-cdo3aet-fgk-fab显示出与111in-do3a-eda-fab为同水平的血中浓度,并且可抑制向肾脏的蓄积,能够得到较低的肾脏-血液比。由上述的实验结果可见,作为连接基团,具有苄基酰胺结构的本发明的化合物111in-cdo3aet-fgk-fab(来源于兔血清igg)、111in-cdo3aet-fgk-fab(来源于抗c-kitigg)、111in-cdota-bn-co-fgk-fab(来源于抗c-kitigg)、111in-do3a-bn-co-fgk-fab(来源于兔血清igg)中,在肾脏中的放射活性得到显著抑制,而作为连接基团,具有硫代脲结构的比较化合物111in-do3a-bn-scn-mvk-fab、及具有亚乙基结构的111in-do3a-eda-fab(来源于兔血清igg)中,在肾脏中的放射活性显示出较高的值。根据上述结果及与bbmvs的孵育试验的试验结果,可以认为,具有苄基酰胺结构的本发明的化合物中,在进行基于酶的代谢、被肾脏摄入之前,放射性金属部位是游离的,而另一方面,具有硫代脲结构的比较化合物中,未被酶识别,可认为没有代谢物的游离。图7a~c示出了111in-cdo3aibu-fgk-fab(来源于抗c-kitigg)、111in-dota-bn-scn-fab(来源于抗c-kitigg)、及111in-cdo3aet-fgk-fab(来源于抗c-kitigg)的结果的比较。由上述的结果可知,111in-cdo3aibu-fgk-fab较之111in-cdo3aet-fgk-fab而言进一步抑制向肾脏的蓄积,能够得到更低的肾脏-血液比。[表3]表3111in-cdo3aet-fgk-fab(来源于兔血清igg)单位为“%id/g”,但是,*为“%id”[表4]表4111in-cdo3aet-fgk-fab(来源于抗c-kitigg)单位为“%id/g”,但是,*为“%id”[表5]表5111in-do3a-eda-fab(来源于兔血清igg)单位为“%id/g”,但是,*为“%id”[表6]表6111in-cdota-bn-co-fgk-fab(来源于抗c-kitigg)单位为“%id/g”,但是,*为“%id”[表7]表7111in-do3a-bn-scn-mvk-fab(来源于兔血清igg)单位为“%id/g”,但是,*为“%id”[表8]表8111in-do3a-bn-co-fgk-fab(来源于兔血清igg)单位为“%id/g”,但是,*为“%id”[表9]表9111in-cdo3aibu-fgk-fab(来源于兔血清igg)单位为“%id/g”,但是,*为“%id”[表10]表10111in-cdo3aibu-fgk-fab(来源于抗c-kitigg)单位为“%id/g”,但是,*为“%id”[表11]表11111in-dota-bn-scn-fab(来源于抗c-kitlgg)单位为“%id/g”,但是,*为“%id”〔尿液中放射活性的分析〕将111in-cdo3aet-fgk-fab(来源于兔血清igg)用d-pbs(-)稀释。经由小鼠尾静脉,施予将fab浓度调节为5μg/100μl的111in-cdo3aet-fgk-fab(来源于兔血清igg)溶液(4μci/100μl/只),将到24小时后为止积聚的尿液用0.45μm的过滤器过滤后,通过se-hplc分析化学形态。此外,通过向回收的尿液中加入2倍量的etoh使蛋白质沉淀,并在以15,000g离心5分钟后回收上清液。回收上清液后,使用66%etoh溶液100μl洗涤颗粒,再次离心并回收上清液,实施2次前述操作,算出放射活性的针对上清液的回收率。然后,将上清液用d-pbs(-)稀释以使得etoh浓度为15%以下,利用rp-hplc进行分析。图8示出了将111in-cdo3aet-fgk-fab(来源于兔血清igg)施予至小鼠后24小时为止排泄的尿液中放射活性的化学形态的分析结果。以上,根据尿液中的放射活性的分析,通过图8a所示的利用se-hplc的分析可以发现,大部分放射活性以小分子级分被排泄,由图8b所示的rp-hplc的结果可知,111in-cdo3aet-fgk-fab中,小分子级分中的主要放射活性为111in-cdo3aet-phe(将111in-cdo3aet-fgk-fab在苯丙氨酸-甘氨酸之间切割而得到的化合物)。将111in-cdo3aet-fgk-fab(来源于兔血清igg)变更为111in-cdota-bn-co-fgk-fab(来源于抗c-kitigg),将施予后6小时后为止排泄的尿液进行积聚并分析,除此以外,通过与上述同样的方法,分析了尿液中放射活性。图9示出了将111in-cdota-bn-co-fgk-fab(来源于抗c-kitigg)施予至小鼠后6小时为止排泄的尿液中放射活性的化学形态的分析结果。由rp-hplc的结果可知,111in-cdota-bn-co-fgk-fab中,小分子级分中的主要放射活性为111in-cdota-phe(将111in-cdota-bn-co-fgk-fab在苯丙氨酸-甘氨酸之间切割而得到的化合物)。将111in-cdo3aet-fgk-fab(来源于兔血清igg)变更为111in-do3a-bn-co-fgk-fab(来源于兔血清igg),除此以外,通过与上述同样的方法,分析了尿液中放射活性。图10示出了将111in-do3a-bn-co-fgk-fab(来源于兔血清igg)施予至小鼠后24小时为止排泄的尿液中放射活性的化学形态的分析结果。由rp-hplc的结果可知,111in-do3a-bn-co-fgk-fab中,小分子级分中的主要放射活性为111in-do3a-phe(将111in-do3a-bn-co-fgk-fab在苯丙氨酸-甘氨酸之间切割而得到的化合物)。将111in-cdo3aet-fgk-fab(来源于兔血清igg)变更为111in-cdo3aibu-fgk-fab(来源于兔血清igg),除此以外,通过与上述同样的方法,分析了尿液中放射活性。图11示出了将111in-cdo3aibu-fgk-fab(来源于兔血清igg)施予至小鼠后24小时为止排泄的尿液中放射活性的化学形态的分析结果。以上,根据尿液中的放射活性的分析,通过图11a所示的利用se-hplc的分析可以发现,大部分的放射活性以小分子级分的形式被排泄,由图11b所示的rp-hplc的结果可知,111in-cdo3aibu-fgk-fab(来源于兔血清igg)中,小分子级分中的主要放射活性为111in-cdo3aibu-phe(将111in-cdo3aibu-fgk-fab在苯丙氨酸-甘氨酸之间切割而得到的化合物)。〔spect/ct成像〕将利用上述的方法制备的111in-cdo3aet-fgk-fab(来源于抗c-kitigg)用d-pbs(-)稀释。经由上述sy皮下肿瘤模型小鼠的尾静脉,施予将fab浓度调节为25μg/100μl的111in-cdo3aet-fgk-fab溶液(45μci/100μl/只)。使用spect/ct装置(spect4ct,trifoilimaging,ca),在开口直径1mm、5孔针孔准直仪360度收集、16次成像、14分钟/成像的条件下,从施予后2.5小时起对各组中的2只小鼠进行成像。将111in-cdo3aet-fgk-fab(来源于抗c-kitigg)溶液(45μci/100μl/只)变更为111in-do3a-bn-scn-mvk-fab(来源于抗c-kitigg)溶液(14μci/100μl/只),除此以外,同样地操作,经由sy皮下肿瘤模型小鼠的尾静脉进行施予,自施予后2.5小时起进行成像。图12为将111in-do3a-bn-scn-mvk-fab(来源于抗c-kitigg)溶液、或111in-cdo3aet-fgk-fab(来源于抗c-kitigg)溶液施予至sy皮下肿瘤模型小鼠后2.5小时的spect/ct成像。在施予2.5小时后,111in-cdo3aet-fgk-fab向肾脏的积聚低,使肿瘤清楚地成像。另一方面,111in-do3a-bn-scn-mvk-fab中,虽然使肿瘤成像,但在肾脏中也观察到高放射活性。以上,放射性标记药物向肾脏的积聚低,能够提高放射线成像诊断的灵敏度、精度。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1