5-甲氧基-7-氮杂吲哚的合成方法与流程
本发明涉及药物中间体领域,具体涉及一种5-甲氧基-7-氮杂吲哚的合成方法。
背景技术:
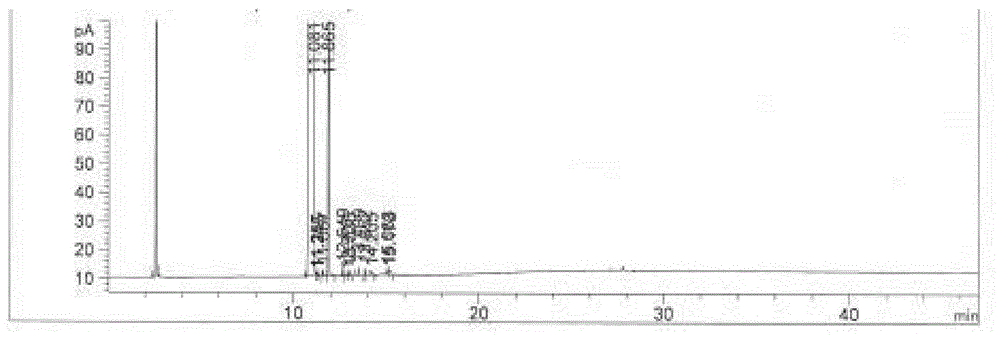
:氮杂吲哚可以看做是吲哚的生物电子等排体,而吲哚分子单元在生命过程中发挥着巨大的作用,很多药物分子都含有吲哚单元,所以氮杂吲哚类化合物在药物活性分子设计和合成方面发挥着重要的作用。氮杂吲哚及其衍生物具有抑制多种蛋白酶的活性。比如天然产物中含7-氮杂吲哚结构的化合物是一种重要的生物碱,具有抗癌、抗糖尿病等生理活性。由于该类化合物特殊的理化性质和药理活性,所以化学家通常以7-氮杂吲哚结构的化合物作为先导化合物进行结构改造。5-甲氧基-7-氮杂吲哚作为制备活性药物成分的关键中间体,目前主要是由5-溴-7-氮杂吲哚制备。其制备为:而作为原料的5-溴-7氮杂吲哚不易获得,制备成本较高。目前关于5-溴-7氮杂吲哚的制备方法主要有以下几种:us20100204214、us20070049615、heterocycle,2003,60(4):865-877、jacs,2006,128(45):14426-14437以及chemmedchem,2007,2(7):1071-1075均公开了采用下述方法制备5-溴-7氮杂吲哚,其是以7-氮杂吲哚为原料,经过溴化、消除、还原、氧化而成,反应方程式如下:该方法步骤长,需大量的溴素参加反应,最后又消去,产生大量废液,操作繁琐,总收率低。wo2003064413,wo2004078757和wo2007135398均公开了以7-氮杂吲哚为原料,经氢化还原、溴化和脱氢得到5-溴-7-氮杂吲哚,其反应路线如下:其缺点是,对于溴化步骤,过量使用溴素,产生大量废液,污染严重;脱氢步骤,采用二氧化锰,产生大量含锰废渣,带来污染。synthesis2008,(13):2049-2054、wo20060183758、wo20080124849、wo2009016460以及us20110028511均公开了5-溴-2-氨基吡啶为原料,经碘化、sonogashira交叉偶联、然后在强碱中水解、环合得到5-溴-7-氮杂吲哚,其反应路线如下:该路线使用多种金属催化剂,交叉偶联副产物不好纯化,条件较为苛刻,工业成本高。中国专利申请号cn201210025316提供了一种5-溴-7-氮杂吲哚的制备方法,以7-氮杂吲哚为原料,主要包含步骤:7-氮杂吲哚与亚硫酸氢钠反应生成二氢-7-氮杂吲哚-2-磺酸钠盐;二氢-7-氮杂吲哚-2-磺酸钠盐发生溴代反应生成二氢-5-溴-7-氮杂吲哚-2-磺酸钠盐;二氢-5-溴-7-氮杂吲哚-2-磺酸钠盐在碱性条件下脱磺酸钠生成5-溴-7-氮杂吲哚,其反应路线如下:该制备方法存在溴素消耗大、溴化过程中交叉偶联副产多的问题,产生大量废液,操作繁琐。wo2011110479和wo2011109932均公开了5-溴-3-甲基-2-氨基吡啶为原料,在四氢吡咯和n,n-二甲基甲酰胺二甲基缩醛(dmf-dma)作用下生成得到中间体,随后中间体利用lda(n,n-二异丙基氨基锂)环合得到5-溴-7-氮杂吲哚,其反应路线为:该方法虽然缩短了合成路线,但是总收率比较低。中国专利申请号cn201410596601.3公开了以2-氨基-3-甲基-5-溴吡啶为原料,在的卡罗酸氧化作用下生成2-硝基-3-甲基-5-溴吡啶,随后在四氢吡咯和n,n-二甲基甲酰胺二甲基缩醛(dmf-dma)作用下生成得到中间体,最后在雷尼镍/85%水合肼体系或其他低价金属下作用下还原关环形成5-溴-7-氮杂吲哚,其反应路线如下:该方法产生大量的废酸,三废压力大,工业成本高。并且,目前报道的制备5-溴-7-氮杂吲哚的工艺主要存在以下缺点:1.涉及的原料或试剂的价格昂贵。2.包含一些对于生产较危险的氧化还原工艺,不利于实现工业化。3.副产物较多,纯化过程损失收率,导致了总收率低。技术实现要素:为此,本发明所解决的技术的问题是:现有技术中采用5-溴-7-氮杂吲哚制备5-甲氧基-7-氮杂吲哚,但是5-溴-7-氮杂吲哚不易获得,生产成本较高,收率低。本发明的目的是提供一种5-甲氧基-7-氮杂吲哚的合成方法,其与现有的制备5-甲氧基-7-氮杂吲哚的方法相比,工艺简单,对生产条件要求低,产物提纯容易且收率高,显著提高了5-甲氧基-7-氮杂吲哚的生产效率和产品质量。本发明提供了一种5-甲氧基-7-氮杂吲哚,其包含下述工序:工序a):以化合物1为原料制备得到化合物2;工序b):通过化合物2制备得到5-甲氧基-7-氮杂吲哚。具体来说,本发明提出了如下技术方案。本发明提供了一种5-甲氧基-7-氮杂吲哚的合成方法,其包含下述工序:工序a):以化合物1为原料制备得到化合物2;工序b):通过化合物2制备得到5-甲氧基-7-氮杂吲哚。优选的,对于上述所述的合成方法,其中,工序b)是将化合物2溶解在第三有机溶剂中,然后加入碱进行反应,接着加入n,n-二甲基甲酰胺形成醛基中间体,最后醛基中间体在酸性条件下环合后,结晶得到产品;其中,优选加入碱对甲基去质子化;较优选的,所述在酸性条件下环合为在酸性淬灭剂条件下进行淬灭以及环合。优选的,对于上述所述的合成方法,其中,工序b)中,所述的碱选自正丁基锂、二异丙基胺基锂、双(三甲基硅基)氨基锂或双(三甲基硅基)氨基钠中的一种,优选为正丁基锂。优选的,对于上述所述的合成方法,其中,工序b)中,所述第三有机溶剂选自于四氢呋喃、乙腈、二氯甲烷、氯仿、四氯化碳、乙醚、甲基丙基醚、正丁醚、乙二醇二甲醚、二乙二醇二甲醚、苯甲醚、甲基叔丁基醚、1,2-二氯乙烷、苯、甲苯、对二甲苯、己烷、庚烷或辛烷中的一种,优选为四氢呋喃;优选的,所述化合物2与所述第三有机溶剂的质量比为1:2~100,优选为1:5~80,较优选为1:8~15。优选的,对于上述所述的合成方法,其中,工序b)中,所述酸性淬灭剂选自于浓盐酸、硫酸、硝酸、醋酸或柠檬酸中的一种,优选为浓盐酸;优选的,所述化合物2与所述酸性淬灭剂的质量比为1:2~10,优选为1:2~5,较优选为1:2.5~3。优选的,对于上述所述的合成方法,其中,工序b)中,所述化合物2与所述碱的摩尔比为1:2.0~6.0,优选为1:2.0~2.5;优选的,所述化合物2与所述n,n-二甲基甲酰胺的摩尔比为1:1.0~10.0,优选为1:1.0~5,较优选为1:1.0~1.5。优选的,对于上述所述的合成方法,其中,工序b)中,加入碱的温度为-40~30℃,优选为-20~0℃;优选的,反应时间为2-12小时;优选的,加入n,n二甲基甲酰胺的温度为-40~25℃,优选为-20~20℃;优选的,反应时间为2-12小时,优选为4-6小时。优选的,对于上述所述的合成方法,其中,工序b)中,所述进行淬灭的反应温度为-10~30℃,优选为0~5℃;优选的,所述环合的反应温度为0~45℃,优选为40~45℃;优选的,所述环合的反应时间为1-6小时。优选的,对于上述所述的合成方法,其中,工序a)包括如下步骤(1)和步骤(2):(1)2-氨基-3-甲基-5-甲氧基吡啶的合成:将化合物1溶解在第一有机溶剂中,在第一催化剂的作用下,化合物1与甲醇盐进行反应,得到化合物2-氨基-3-甲基-5-甲氧基吡啶;(2)化合物2的合成:将化合物2-氨基-3-甲基-5-甲氧基吡啶与氨基保护剂反应得到化合物2。优选的,对于上述所述的合成方法,其中,所述步骤(2)中,将化合物2-氨基-3-甲基-5-甲氧基吡啶溶解在第二有机溶剂中,然后在第二催化剂的存在下,与氨基保护剂和碱进行反应得到氨基保护的化合物2,其中化合物2中,r为氨基保护基;优选在与氨基保护剂和碱反应之后,进一步加入淬灭剂进行淬灭,重结晶得到氨基保护的化合物2。优选的,对于上述所述的合成方法,其中,在步骤(1)中,所述第一有机溶剂选自于甲醇、乙醇、乙二醇、丙醇、异丙醇、丙三醇、甲基乙二醇,1-丁醇、2-丁醇、2-甲基-1-丙醇、2-甲基-2-丙醇、甲基丙三醇、2-甲基丙三醇和二甲基乙二醇中的一种或两种以上,优选为甲醇;优选的,所述化合物1与所述第一有机溶剂的质量比为1:0.5~50,优选为1:3~10。优选的,对于上述所述的合成方法,其中,在步骤(1)中,所述第一催化剂选自于金属铜、亚铜盐或铜盐中的一种;优选的,所述第一催化剂选自于铜粉、溴化亚铜、氧化亚铜、氧化铜或硫酸铜中的一种。优选的,对于上述所述的合成方法,其中,在步骤(1)中,所述化合物1与所述第一催化剂的摩尔比为1:0.05~2,优选为1:0.1~1.5,较优选为1:0.1~0.5;优选的,所述甲醇盐选自于甲醇钠、甲醇钾或甲醇锂中的一种,优选为甲醇钠;优选的,所述化合物1与所述甲醇盐的摩尔比为1:0.8~20,优选为1:1~15,优选为1:1.5~3.0。优选的,对于上述所述的合成方法,其中,在步骤(1)中,所述反应的温度为80~150℃,优选为100~130℃,较优选为120~130℃;优选的,所述反应的时间为1~20小时,优选为1~15小时,较优选为3~8小时。优选的,对于上述所述的合成方法,其中,在步骤(2)中,所述第二有机溶剂选自于四氢呋喃、乙腈、甲醇、乙醇、乙二醇、丙醇、异丙醇、丙三醇、甲基乙二醇,1-丁醇、2-丁醇、2-甲基-1-丙醇、2-甲基-2-丙醇、甲基丙三醇、2-甲基丙三醇、二甲基乙二醇和叔丁醇中的一种或两种以上,优选为叔丁醇;优选的,所述化合物2-氨基-3-甲基-5-甲氧基吡啶与所述第二有机溶剂的质量比为1:0.5~30.0,优选为1:1~20,较优选为1:1.0~5.0。优选的,对于上述所述的合成方法,其中,在步骤(2)中,所述第二催化剂选自于吡啶、n,n-二异丙基乙胺、四甲基乙二胺、三丙基胺、4-二甲氨基吡啶、三亚乙基二胺、四丁基氢氧化铵、γ-氯丙基甲基二甲氧基硅烷、n-甲基吗啉和三乙胺中的一种或两种以上,优选为4-二甲氨基吡啶;优选的,所述化合物2-氨基-3-甲基-5-甲氧基吡啶和所述第二催化剂的摩尔比为1:0.01~1.0,优选为1:0.02~0.2,较优选为1:0.02~0.05。优选的,对于上述所述的合成方法,其中,在步骤(2)中,所述氨基保护剂为二碳酸二叔丁酯;优选的,反应时,所述氨基保护剂以熔融成液态形式或溶解在第四有机溶剂中形成溶液的形式滴加;优选的,所述化合物2-氨基-3-甲基-5-甲氧基吡啶与氨基保护剂的摩尔比为1:1~10,优选为1:1.1~5,较优选为1:2.1-2.5;优选的,所述第四有机溶剂选自于四氢呋喃、乙腈、甲醇、乙醇、乙二醇、丙醇、异丙醇、丙三醇、甲基乙二醇,1-丁醇、2-丁醇、2-甲基-1-丙醇、2-甲基-2-丙醇、甲基丙三醇、2-甲基丙三醇、二甲基乙二醇和叔丁醇中的一种或两种以上,优选为叔丁醇;优选的,所述氨基保护剂与所述第四有机溶剂的质量比为1:0.5~30,优选为1:1~20,较优选为1:0.5~4。优选的,对于上述所述的合成方法,其中,在步骤(2)中,所述碱选自于碱金属或碱土金属的氢氧化物、碱金属或碱土金属的碳酸盐、碱金属的烷基盐、碱金属的二异丙基胺化盐或双三甲基硅基氨基盐中的一种,优选为氢氧化钠、氢氧化钾、正丁基锂、仲丁基锂、异丁基锂、叔丁基锂或双三甲基硅基氨基钠中的一种;优选的,所述化合物2-氨基-3-甲基-5-甲氧基吡啶与所述碱的摩尔比为1:1.0~5.0,优选为1:1.5~2.5。优选的,对于上述所述的合成方法,其中,在步骤(2)中,所述反应温度为-40℃~85℃,优选为60~85℃;优选的,加入氨基保护剂后的反应时间为1~40小时,优选为6-8小时;优选的,加入碱后的反应时间为1~40小时,优选为6~10小时。优选的,对于上述所述的合成方法,其中,所述淬灭剂选自于盐酸、硫酸、硝酸、水、甲酸、醋酸或柠檬酸中的一种。本发明所取得的有益效果是:本发明所述的制备方法,与目前报道的合成方法相比大大缩短了合成路线;所用原料为2-氨基-3-甲基-5-溴吡啶,与现有工艺的5-溴-7-氮杂吲哚相比,原料廉价易得;该方案有效避免了现有技术的副产物,总收率提高,所得产品纯度较高,并且本发明所用原料以及试剂均为廉价易得,对设备要求低,非常易于实现工业化。附图说明图1是实施例一步骤(1)所得到的化合物的gc谱图。图2是实施例一步骤(1)所得到的化合物的gc-ms谱图。图3是实施例一所得到的化合物2的hplc谱图。图4是实施例一所得到的化合物2的lc-ms谱图图5是实施例一所得到的化合物2的1hnmr谱图。图6是实施例一所得到的产品的hplc谱图。图7是实施例一所得到的产品的lc-ms谱图图8是实施例一所得到的产品的1hnmr谱图。具体实施方式如上所述,本发明提供了一种5-甲氧基-7-氮杂吲哚的合成方法,其包含下述工序:工序a):以化合物1为原料制备得到化合物2;工序b):通过化合物2制备得到5-甲氧基-7-氮杂吲哚。在本发明优选的一种具体实施方式中,其中,工序b)是将化合物2溶解在第三有机溶剂中,然后加入碱进行反应,接着加入n,n-二甲基甲酰胺形成醛基中间体,最后醛基中间体在酸性条件下环合后,结晶得到产品;其中,优选加入碱对甲基去质子化;较优选的,所述在酸性条件下环合为在酸性淬灭剂条件下进行淬灭以及环合。在本发明优选的一种具体实施方式中,其中,工序b)中,所述碱选自正丁基锂、二异丙基胺基锂、双(三甲基硅基)氨基锂、或双(三甲基硅基)氨基钠中的一种;优选为正丁基锂。在本发明优选的一种具体实施方式中,其中,工序b)中,所述第三有机溶剂选自于四氢呋喃、乙腈、二氯甲烷、氯仿、四氯化碳、乙醚、甲基丙基醚、正丁醚、乙二醇二甲醚、二乙二醇二甲醚、苯甲醚、甲基叔丁基醚、1,2-二氯乙烷、苯、甲苯、对二甲苯、己烷、庚烷或辛烷中的一种,优选为四氢呋喃;优选的,所述化合物2与所述第三有机溶剂的质量比为1:2~100,优选为1:5~80,较优选为1:8~15。在本发明优选的一种具体实施方式中,其中,所述酸性淬灭剂选自于浓盐酸、硫酸、硝酸、醋酸或柠檬酸中的一种,优选为浓盐酸;优选的,所述化合物2与所述酸性淬灭剂的质量比为1:2~10,优选为1:2~5,较优选为1:2.5~3;优选的,所述浓盐酸是指质量分数为25-35%的盐酸。在本发明优选的一种具体实施方式中,其中,工序b)中,所述化合物2与所述碱的摩尔比为1:2.0~6.0,优选为1:2.0~2.5;优选的,所述化合物2与所述n,n-二甲基甲酰胺的摩尔比为1:1.0~10.0,优选为1:1.0~5,较优选为1:1.0~1.5。在本发明优选的一种具体实施方式中,其中,工序b)中,加入碱的温度为-40~30℃,优选为-20~0℃;优选的,反应时间为2-12小时;优选的,加入n,n二甲基甲酰胺的温度为-40~25℃,优选为-20~20℃;优选的,反应时间为2-12小时,优选为4-6小时。在本发明优选的一种具体实施方式中,其中,工序b)中,所述进行淬灭的反应温度为-10~30℃,优选为0~5℃;优选的,所述环合的反应温度为0~45℃,优选为40~45℃;优选的,所述环合的反应时间为1-6小时。在本发明优选的一种具体实施方式中,其中,工序a)包括如下步骤(1)和步骤(2):(1)2-氨基-3-甲基-5-甲氧基吡啶的合成:将化合物1溶解在第一有机溶剂中,在第一催化剂的作用下,化合物1与甲醇盐进行反应,得到化合物2-氨基-3-甲基-5-甲氧基吡啶;(2)化合物2的合成:将化合物2-氨基-3-甲基-5-甲氧基吡啶与氨基保护剂反应得到化合物2。在本发明优选的一种具体实施方式中,其中,所述步骤(2)中,将化合物2-氨基-3-甲基-5-甲氧基吡啶溶解在第二有机溶剂中,然后在第二催化剂的存在下,与氨基保护剂和碱进行反应,得到氨基保护的化合物2,其中化合物2中,r为氨基保护基;优选在与氨基保护剂和碱反应之后,进一步加入淬灭剂进行淬灭,重结晶得到氨基保护的化合物2;优选的,化合物2-氨基-3-甲基-5-甲氧基吡啶可以先与氨基保护剂进行反应,然后再与碱进行反应得到化合物2,也可以是化合物2-氨基-3-甲基-5-甲氧基吡啶先与碱进行反应,然后再与氨基保护剂进行反应得到化合物2。在本发明优选的一种具体实施方式中,其中,在步骤(1)中,所述第一有机溶剂选自于甲醇、乙醇、乙二醇、丙醇、异丙醇、丙三醇、甲基乙二醇,1-丁醇、2-丁醇、2-甲基-1-丙醇、2-甲基-2-丙醇、甲基丙三醇、2-甲基丙三醇和二甲基乙二醇中的一种或两种以上,优选为甲醇;优选的,所述化合物1与所述第一有机溶剂的质量比为1:0.5~50,优选为1:3~10。在本发明优选的一种具体实施方式中,其中,在步骤(1)中,所述第一催化剂选自于金属铜、亚铜盐或铜盐中的一种;优选的,所述第一催化剂选自于铜粉、溴化亚铜、氧化亚铜、氧化铜或硫酸铜中的一种;优选的,所述化合物1与所述第一催化剂的摩尔比为1:0.05~2.0,优选为1:0.1~1.5,较优选为1:0.1~0.5;优选的,所述甲醇盐选自于甲醇钠、甲醇钾或甲醇锂中的一种,优选为甲醇钠;优选的,所述化合物1与所述甲醇盐的摩尔比为1:0.8~20,优选为1:1~15,较优选为1:1.5~3.0。在本发明优选的一种具体实施方式中,其中,在步骤(1)中,所述反应的温度为80~150℃,优选为100~130℃,较优选为120~130℃;优选的,所述反应的时间为1~20小时,优选为1~15小时,较优选为3~8小时。在本发明优选的一种具体实施方式中,其中,在步骤(2)中,所述第二有机溶剂选自于四氢呋喃、乙腈、甲醇、乙醇、乙二醇、丙醇、异丙醇、丙三醇、甲基乙二醇,1-丁醇、2-丁醇、2-甲基-1-丙醇、2-甲基-2-丙醇、甲基丙三醇、2-甲基丙三醇、二甲基乙二醇和叔丁醇中的一种或两种以上,优选为叔丁醇;优选的,所述化合物2-氨基-3-甲基-5-甲氧基吡啶与所述第二有机溶剂的质量比为1:0.5~30.0,优选为1:1~20,较优选为1:1.0~5.0。在本发明优选的一种具体实施方式中,其中,在步骤(2)中,所述第二催化剂选自于吡啶、n,n-二异丙基乙胺、四甲基乙二胺、三丙基胺、4-二甲氨基吡啶、三亚乙基二胺、四丁基氢氧化铵、γ-氯丙基甲基二甲氧基硅烷、n-甲基吗啉和三乙胺中的一种或两种以上,优选为4-二甲氨基吡啶;优选的,所述化合物2-氨基-3-甲基-5-甲氧基吡啶和所述第二催化剂的摩尔比为1:0.01~1:1.0,优选为1:0.02~0.2,较优选为1:0.02~0.05。在本发明优选的一种具体实施方式中,其中,在步骤(2)中,所述氨基保护剂为二碳酸二叔丁酯;优选的,反应时,所述氨基保护剂以熔融成液态形式或溶解在第四有机溶剂中的形式滴加;优选的,所述化合物2-氨基-3-甲基-5-甲氧基吡啶与氨基保护剂的摩尔比为1:1~10,优选为1:1.1~5,较优选为1:2.1-2.5;优选的,所述第四有机溶剂选自于四氢呋喃、乙腈、甲醇、乙醇、乙二醇、丙醇、异丙醇、丙三醇、甲基乙二醇,1-丁醇、2-丁醇、2-甲基-1-丙醇、2-甲基-2-丙醇、甲基丙三醇、2-甲基丙三醇、二甲基乙二醇和叔丁醇中的一种或两种以上,优选为叔丁醇;优选的,所述氨基保护剂与所述第四有机溶剂的质量比为1:0.5~30,优选为1:1~20,较优选为1:0.5~4。在本发明优选的一种具体实施方式中,其中,在步骤(2)中,所述碱选自于碱金属或碱土金属的氢氧化物、碱金属或碱土金属的碳酸盐、碱金属的烷基盐、碱金属的二异丙基胺化盐或双三甲基硅基氨基盐中的一种,优选为氢氧化钠、氢氧化钾、正丁基锂、仲丁基锂、异丁基锂、叔丁基锂或双三甲基硅基氨基钠中的一种;优选的,所述化合物2-氨基-3-甲基-5-甲氧基吡啶与所述碱的摩尔比为1:1.0~5.0,优选为1:1.5~2.5。在本发明优选的一种具体实施方式中,其中,在步骤(2)中,所述反应温度为-40℃~85℃,优选为60~85℃;优选的,加入氨基保护剂后的反应时间为1~40小时;优选的,加入碱后的反应时间为1~40小时,优选为6~10小时;优选的,所述加入氨基保护剂后的反应温度为-40℃~85℃,优选为60~85℃,所述加入碱后的反应温度为-40℃~85℃,优选为60~85℃。在本发明优选的一种具体实施方式中,其中,所述淬灭剂选自于盐酸、硫酸、硝酸、水、甲酸、醋酸或柠檬酸中的一种。下面的实施例是对发明的合成方法做进一步的详细说明,其中实施例中所使用的化学物质均为常规试剂的化学纯级别。实施例一5-甲氧基-7-氮杂吲哚的合成其合成路线为:(1)化合物2-氨基-3-甲基-5-甲氧基吡啶的合成:将400.0g(2.14mol,1.0eq)的化合物1和2kg的甲醇加入到5l高压釜中,搅拌至固体全部溶解后,依次加入346.6g(6.42mol,3.0eq)的甲醇钠和68.0g(1.07mol,0.5eq)的铜粉,加热至130℃反应8小时,降温至25℃,将釜内物料过滤,滤饼以少量甲醇淋洗,母液浓缩至断流,向其中加入二氯甲烷和水萃取分层,水相以二氯甲烷再萃取一次,合并有机相浓缩回收溶剂,粗品通过40cm填料柱进行减压精馏,收集t=85-90℃/p=2-3mmhg馏分,共计获得153.5g的化合物,纯度98.4%,收率52%;将上述所得到的化合物的结构确认采用gc(agilent7820a)进行确认,其结果如图1所示,其各个峰的保留时间、峰面积及峰高如下表所示:将步骤(1)所得到的化合物采用gc-ms进行结构确认,其结果如图2所示,其中,使用的设备为agilentgc7890,ms5975色谱柱为db-5ms,30m×0.25mm,膜厚度0.25μm载气:he柱流量:0.7ml/min不变进样口温度:280℃,离子源温度:230℃ms温度:150℃,aux-2温度:200℃分流比:100:1柱箱温度:70℃保持0min,以10℃/min到280℃,保温10min100mg样品溶解在1ml乙腈中,注射0.1μl,记录谱图,将碎片与标准比较得到;从图1和图2可以看出,所得到的化合物为2-氨基-3-甲基-5-甲氧基吡啶;(2)化合物2的合成:在40℃,向500ml四口瓶加入20.7g(0.15mol,1.0eq)步骤(1)所得到的化合物2-氨基-3-甲基-5-甲氧基吡啶和82.9g叔丁醇,待物料全部溶解后加入1.1g(0.007mol,0.05eq)4-二甲氨基吡啶,升温至80-85℃,将81.8g(0.38mol,2.5eq)二碳酸二叔丁酯溶解在82.9g的叔丁醇中,并保持四口瓶内温度80-85℃滴加,滴加完毕后在该温度下继续反应6小时得到中间体化合物,然后加入12.0g氢氧化钠(0.30mol,2.0eq),并且在该温度继续反应,反应时间为6小时,反应结束后,使用醋酸淬灭反应液至中性,升温至80-85℃蒸除溶剂至断流、以四氢呋喃萃取2次,然后合并有机相并蒸干溶剂后进行结晶,得到34.0g化合物2,收率95%;将所得到的化合物2的结构采用hplc进行确认,其谱图如图3所示,其中,保留时间、峰宽、峰面积、峰高以及峰面积%如下表所示:峰#保留时间(min)峰宽(min)峰面积(mau*s)峰高(mau)峰面积(%)13.0540.05965.945971.521660.205923.3890.04828.74662e-12.81860e-10.030334.5820.05351.892925.32736e-10.065546.4220.05102876.74316861.5421199.607556.7970.05582.053895.72743e-10.071167.8800.07365.68572e-11.19322e-10.0197总量2888.07918864.57043100%从图3可以看出,所得到的化合物2的纯度为99.6%。将所得到的化合物2采用lc-ms进行结构确认,其谱图如图4所示,其中,流动相a为0.1%甲酸水溶液,其制备方法为1ml甲酸溶于1000ml水中,混匀;流动相b为乙腈,其按照下述方式进行梯度洗脱,色谱柱为安捷伦poroshell120ec-c18,4μm,150×4.6mm(货号:693970-902),检测波长为220nm,柱温为30℃,流速为1.0ml/min,电压为150v,气体温度为325℃,干燥气流为11l/min,进样量为0.5微升。时间(min)流动相a(v/v%)流动相b(v/v%)095510109013109013.195516955从图4可以看出,所得到的化合物2为2-叔丁氧基氨基-3-甲基-5-甲氧基吡啶。将所得到的化合物2的结构确认采用h1-nmr(bruker400mhz)进行确认,其结果如图5所示;从图5可以看出,所得到的化合物2为2-叔丁氧基氨基-3-甲基-5-甲氧基吡啶,图中的93.26%是采用核磁内标法测定的化合物2的含量,内标物为1,3,5-三甲氧基苯;(3)产品的合成向2l四口瓶通入氮气保护,加入72.0g(0.3mol,1.0eq)步骤(2)所得到的化合物2和720ml的四氢呋喃,降温至-10±5℃,并控制在该温度下滴加204.0g(0.75mol,2.5eq)2.5m正丁基锂的己烷溶液,滴加完毕后在该温度继续反应2.5小时,然后在该温度下滴加26.3g(0.36mol,1.2eq)的n,n-二甲基甲酰胺,滴加完毕后,保持-10±5℃搅拌6小时至反应完全,将瓶中混合物保持0±5℃慢慢倾倒入216g的35%浓盐酸中,并保持0±5℃淬灭反应,将反应液升温至40-45℃反应2小时,待反应结束后分层除去有机层,将水相以氢氧化钠中和至ph=11~12,然后用四氢呋喃萃取两次,合并的有机相以饱和食盐水洗涤一次后,合并有机相回收溶剂后进行结晶得到35.3g产品,收率79%;将所得到的产品的结构采用hplc进行确认,其谱图如图6所示,其中,保留时间、峰宽、峰面积、峰高以及峰面积%如下表所示:峰#保留时间(min)峰宽(min)峰面积(mau*s)峰高(mau)峰面积(%)13.1060.05776.415561.713320.099823.7930.05513.608131.024320.056135.1890.065214.622343.207280.227545.7710.05246396.519531943.4635099.528956.7270.06505.632821.240690.0876总量6426.798381950.64911100%从图6可以看出,所得到的产品的纯度为99.5%。将所得到的产品采用lc-ms进行结构确认,其谱图如表7所示,其中,流动相a为0.1%甲酸水溶液,其制备方法为1ml甲酸溶于1000ml水中,混匀;流动相b为乙腈,其按照下述方式进行梯度洗脱,色谱柱为安捷伦poroshell120ec-c18,4μm,150×4.6mm(货号:693970-902),检测波长为220nm,柱温为30℃,流速为1.0ml/min,电压为150v,气体温度为325℃,干燥气流为11l/min,进样量为0.5微升。时间(min)流动相a(v/v%)流动相b(v/v%)095510109013109013.195516955从图7可以看出,所得到的产品为5-甲氧基-7-氮杂吲哚。将所得到的产品的结构确认采用h1-nmr进行确认,其结果如图8所示;从图8可以看出,所得到的产品为5-甲氧基-7-氮杂吲哚,图中的94.88%是采用核磁内标法测定的产品的含量,内标物为1,3,5-三甲氧基苯。实施例二5-甲氧基-7-氮杂吲哚的合成(1)化合物2-氨基-3-甲基-5-甲氧基吡啶的合成:将200.0g(1.07mol,1.0eq)的化合物1和1kg的甲醇加入到2l高压釜中,搅拌至固体全部溶解后,依次加入173.4g(3.21mol,3.0eq)的甲醇钠和76.8g(0.53mol,0.5eq)的溴化亚铜,加热至130℃反应20小时,降温至25℃,将釜内物料过滤,滤饼以少量甲醇淋洗,母液浓缩至断流,向其中加入二氯甲烷和水萃取分层,水相用二氯甲烷再萃取一次,合并有机相浓缩至断流,粗品通过40cm填料柱进行减压精馏,收集t=85-90℃/p=2-3mmhg馏分,共计获得43.0g的化合物,纯度98.2%,收率29%;采用实施例一相同的方法对所得到的化合物进行分析,经确认,所得到的化合物为2-氨基-3-甲基-5-甲氧基吡啶;(2)化合物2的合成:在0℃,向2l四口瓶加入20.7g(0.15mol,1.0eq)步骤(1)所得到的化合物2-氨基-3-甲基-5-甲氧基吡啶和621g四氢呋喃,待物料全部溶解后加入0.12g(0.0015mol,0.01eq)吡啶,降温至-10℃,将81.8g(0.38mol,2.5eq)二碳酸二叔丁酯溶解在82.9g的四氢呋喃中,并保持四口瓶内温度-10-0℃滴加,滴加完毕后在该温度下继续反应8小时得到中间体化合物,然后加入12.0g氢氧化钠(0.30mol,2.0eq),并且在该温度继续反应,反应时间为6小时,反应结束后,以25%盐酸淬灭反应液至中性,分出有机层,然后合并有机相并蒸干溶剂后进行结晶,得到18.2g化合物2,纯度94.3%,收率51%;采用实施例一相同的方法对所得到的化合物2进行确认,经分析所得到的化合物2为2-叔丁氧基氨基-3-甲基-5-甲氧基吡啶;(3)产品的合成向1l四口瓶通入氮气保护,加入22.7g(0.096mol,1.0eq)步骤(2)所得到的化合物2和340.5ml的四氢呋喃,降温至-40±5℃,并控制在该温度下滴加52.0g(0.192mol,2.0eq)2.5m正丁基锂的己烷溶液,滴加完毕后,保持-40±5℃继续反应2小时,然后在该温度下滴加10.5g(0.144mol,1.5eq)的n,n-二甲基甲酰胺,滴加完毕后,保持-40±5℃搅拌20小时,将瓶中混合物保持0±5℃慢慢倾倒入45.4g的35%浓盐酸中淬灭反应,并保持0±5℃淬灭反应,将反应液升温至40-45℃反应2小时,待反应结束后分层除去有机层,将水相以氢氧化钠中和至ph=11~12,然后用四氢呋喃萃取两次,合并的有机相以饱和食盐水洗涤一次后,合并有机相回收溶剂后进行结晶得到10.0g产品,纯度95.6%,收率71%;采用与实施例一相同的方法对所得到的产品进行结构确认,经确认所得到的产品为5-甲氧基-7-氮杂吲哚。实施例三5-甲氧基-7-氮杂吲哚的合成(1)化合物2-氨基-3-甲基-5-甲氧基吡啶的合成:将200.0g(1.07mol,1.0eq)的化合物1和1kg的甲醇加入到2l高压釜中,搅拌至固体全部溶解后,依次加入225.1g(3.21mol,3.0eq)的甲醇钾和42.6g(0.54mol,0.5eq)的硫酸铜,加热至130℃反应20小时,降温至25℃,将釜内物料过滤,滤饼以少量甲醇淋洗,母液浓缩至断流,向其中加入二氯甲烷和水萃取分层,水相用二氯甲烷再萃取一次,合并有机相浓缩至断流,粗品通过40cm填料柱进行减压精馏,收集t=85-90℃/p=2-3mmhg馏分,共计获得67.6g的化合物,纯度98.1%,收率46%;采用与实施例一相同的方法对所得到的化合物进行结构确认,经确认所得到的化合物为2-氨基-3-甲基-5-甲氧基吡啶。(2)化合物2的合成:在0℃,向500ml四口瓶加入20.7g(0.15mol,1.0eq)步骤(1)所得到的化合物2-氨基-3-甲基-5-甲氧基吡啶和20.7g乙腈,待物料全部溶解后加入1.8g(0.015mol,0.1eq)4-二甲氨基吡啶,升温至20-25℃,将35.5g(0.165mol,1.1eq)二碳酸二叔丁酯溶解在142g的四氢呋喃中,并保持四口瓶内温度20-25℃滴加,滴加完毕后在该温度下继续反应8小时得到中间体化合物,然后加入21g氢氧化钾(0.375mol,2.5eq),并且在该温度继续反应,反应时间为8小时,反应结束后,以25%盐酸淬灭反应液至中性,分出有机层,然后合并有机相并蒸干溶剂后进行结晶,得到8.6g化合物2,纯度95.0%,收率24%;采用与实施例一相同的方法对所得到的化合物2进行结构确认,经确认所得到的化合物2为2-叔丁氧基氨基-3-甲基-5-甲氧基吡啶。(3)产品的合成向1l四口瓶通入氮气保护,加入22.7g(0.096mol,1.0eq)化合物2和181.6ml的四氢呋喃,降温至-10±5℃,并控制在该温度下滴加155.9g(0.576mol,6.0eq)2.5m二异丙基胺基锂的己烷溶液,滴加完毕后继续在该温度下反应3小时,然后在该温度下滴加7.0g(0.096mol,1eq)的n,n-二甲基甲酰胺,滴加完毕后,保持-10±5℃搅拌7小时,将瓶中混合物保持0±5℃慢慢倾倒入56.8g的25%盐酸中全程保持0±5℃淬灭反应,淬灭结束后升温至40-45℃反应2.5小时,反应结束后分层除去有机层,将水相以氢氧化钠中和至ph=11~12,然后用四氢呋喃萃取两次,合并的有机相以饱和食盐水洗涤一次后,合并有机相回收溶剂后进行结晶得到7.9g产品,纯度96.1%,收率56%。采用与实施例一相同的方法对所得到的产品进行结构确认,经确认所得到的产品为5-甲氧基-7-氮杂吲哚。实施例四5-甲氧基-7-氮杂吲哚的合成(1)化合物2-氨基-3-甲基-5-甲氧基吡啶的合成:将40.0g(0.21mol,1.0eq)的化合物1和2000g的乙醇加入到5l高压釜中,搅拌至固体全部溶解后,依次加入12.0g(0.315mol,1.5eq)的甲醇锂和3.0g(0.021mol,0.1eq)的溴化亚铜,加热至140-150℃反应8小时,降温至25℃,将釜内物料过滤,滤饼以少量甲醇淋洗,母液浓缩至断流,向其中加入二氯甲烷和水萃取分层,水相用二氯甲烷再萃取一次,合并有机相浓缩至断流,粗品通过40cm填料柱进行减压精馏,收集t=85-90℃/p=2-3mmhg馏分,共计获得8.3g的化合物,纯度98.2%,收率28%;采用实施例一相同的方法对所得到的化合物进行结构确认,经确认所得到的化合物为2-氨基-3-甲基-5-甲氧基吡啶;(2)化合物2的合成向500ml四口瓶加入37.3g(0.27mol,1.0eq)步骤(1)所得到的化合物2-氨基-3-甲基-5-甲氧基吡啶和187.0g四氢呋喃,待物料全部溶解后于-5℃向反应液中滴加110.2g(0.41mol,1.5eq)2.5m的正丁基锂的己烷溶液,-5℃反应1-2小时后,将123.1g(0.57mol,2.1eq)二碳酸二叔丁酯于-5℃滴加到反应液中,并保持-5℃反应3小时,将反应混合物用水淬灭,过滤收集沉淀物,用水洗涤滤饼,然后真空干燥,得到33.5g的化合物2,纯度92.8%,收率52%;采用与实施例一相同的方法对所得到的化合物2进行结构确认,经确认所得到的化合物2为2-叔丁氧基氨基-3-甲基-5-甲氧基吡啶;(3)产品的合成向500ml四口瓶通入氮气保护,加入22.7g(0.096mol,1.0eq)步骤(2)所得到的化合物2和113.5ml的四氢呋喃,降温至-10±5℃,并控制在该温度下滴加65g(0.24mol,2.5eq)2.5m正丁基锂的己烷溶液,滴加完毕后在该温度继续反应3小时,然后在该温度下滴加33.2g(0.48mol,5eq)的n,n-二甲基甲酰胺,滴加完毕后,保持-10±5℃搅拌6小时,将瓶中混合物保持0±5℃慢慢倾倒入136.2g的25%盐酸中,全程保持0±5℃淬灭反应,然后将淬灭后的反应液升温至40-45℃反应3.5小时,反应结束后分层除去有机层,将水相以氢氧化钠中和至ph=7-8,过滤得到7.8g产品,纯度98.5%,收率55%。采用与实施例一相同的方法对所得到的产品进行结构确认,经确认所得到的产品为5-甲氧基-7-氮杂吲哚。实施例五5-甲氧基-7-氮杂吲哚的合成(1)化合物2-氨基-3-甲基-5-甲氧基吡啶的合成:将40.0g(0.21mol,1.0eq)的化合物1和200g的甲醇加入到500ml高压釜中,搅拌至固体全部溶解后,依次加入34.7g(0.64mol,3.0eq)的甲醇钠和7.2g(0.11mol,0.5eq)的溴化亚铜,加热至100-110℃反应20小时,降温至25℃,将釜内物料过滤,滤饼以少量甲醇淋洗,母液浓缩至断流,向其中加入二氯甲烷和水萃取分层,水相用二氯甲烷再萃取一次,合并有机相浓缩至断流,粗品通过40cm填料柱进行减压精馏,收集t=85-90℃/p=2-3mmhg馏分,共计获得8.9g的化合物,纯度97.6%,收率30%。采用与实施例一相同的方法对所得到的化合物进行结构确认,经确认,所得到的化合物为2-氨基-3-甲基-5-甲氧基吡啶;(2)化合物2的合成:向500ml四口瓶加入37.3g(0.27mol,1.0eq)步骤(1)所得到的化合物2-氨基-3-甲基-5-甲氧基吡啶和187.0g四氢呋喃,待物料全部溶解后于-5℃向反应液中滴加161.6g(0.59mol,2.2eq)2.5m的正丁基锂的己烷溶液,-5℃反应1-2小时后,将73.7g(0.34mol,1.25eq)二碳酸二叔丁酯于-5℃加入到反应液中,并保持-5℃反应3小时,将反应混合物用柠檬酸淬灭,过滤收集沉淀物,用水洗涤滤饼,在有机溶剂中结晶,得到28.3g的化合物2,纯度97.8%,收率44%;采用与实施例一相同的方法对所得到的化合物2进行结构确认,经确认所得到的化合物2为2-叔丁氧基氨基-3-甲基-5-甲氧基嘧啶;(3)产品的合成向250ml四口瓶通入氮气保护,加入22.7g(0.096mol,1.0eq)步骤(2)所得到的化合物2和68ml的四氢呋喃,降温至-10±5℃,并控制在该温度下滴加65g(0.24mol,2.5eq)2.5m正丁基锂的己烷溶液,滴加完毕后在该温度下继续反应3小时,然后在该温度下滴加8.3g(0.12mol,1.2eq)的n,n-二甲基甲酰胺,滴加完毕后,保持-10±5℃反应4小时,将瓶中混合物保持0±5℃慢慢倾倒入68.3g的35%盐酸中淬灭反应,全程保持0±5℃淬灭反应,然后将淬灭后的反应液升温至40-45℃反应1.5小时,反应结束后分层除去有机层,将水相以氢氧化钠中和至ph=11~12,然后用四氢呋喃萃取两次,合并的有机相以饱和食盐水洗涤一次后,合并有机相回收溶剂后进行结晶得到1.7g产品,纯度89.1%,收率12%;采用与实施例一相同的方法对所得到的产品进行结构确认,经确认所得到的产品为5-甲氧基-7-氮杂吲哚。实施例六5-甲氧基-7-氮杂吲哚的合成(1)化合物2-氨基-3-甲基-5-甲氧基吡啶的合成:将40.0g(0.21mol,1.0eq)的化合物1和200g的甲醇加入到500ml高压釜中,搅拌至固体全部溶解后,依次加入170g(3.15mol,15eq)的甲醇钠和5.1g(0.06mol,0.3eq)的氧化铜,加热至140-150℃反应8小时,降温至25℃,将釜内物料过滤,滤饼以少量甲醇淋洗,母液浓缩至断流,向其中加入二氯甲烷和水萃取分层,水相用二氯甲烷再萃取一次,合并有机相浓缩至断流,粗品通过40cm填料柱进行减压精馏,收集t=85-90℃/p=2-3mmhg馏分,共计获得12.1g的化合物,纯度96.9%,收率41%;采用与实施例一相同的方法对所得到的化合物进行结构确认,经确认所得到的化合物为2-氨基-3-甲基-5-甲氧基吡啶;(2)化合物2的合成将9.4g(0.05mol,1.0eq)步骤(1)所得到的化合物2-氨基-3-甲基-5-甲氧基吡啶加入到47.0g四氢呋喃中,配制成溶液a备用;向250ml四口瓶中通入氮气保护,加入22.1g(0.12mol,2.4eq)2.0m双三甲基硅基氨基钠(nahmds)四氢呋喃溶液,于-5℃向反应液中滴加溶液a,并保持-5℃反应1-2小时,将4.4g(0.06mol,1.1eq)二碳酸二叔丁酯于-5℃加入到反应液中,并保持-5℃反应3小时,将反应混合物用水淬灭,过滤收集沉淀物,用水洗涤滤饼,然后真空干燥,得到9.7g的化合物2,纯度98.2%,收率68%;采用与实施例一相同的方法对所得到的化合物2进行结构确认,经确认所得到的化合物2为2-叔丁氧基氨基-3-甲基-5-甲氧基吡啶;(3)产品的合成向500ml四口瓶通入氮气保护,加入22.7g(0.096mol,1.0eq)步骤(2)所得到的化合物2和136ml的四氢呋喃,降温至-20±5℃,并控制在该温度下滴加65g(0.24mol,2.5eq)2.5m正丁基锂的己烷溶液,滴加完毕后在该温度下继续反应4小时,然后在该温度下滴加8.3g(0.12mol,1.2eq)的n,n-二甲基甲酰胺,滴加完毕后,保持-20±5℃反应10小时,将瓶中混合物保持0±5℃慢慢倾倒入68.3g的35%盐酸中淬灭反应,全程保持0±5℃淬灭反应,然后将淬灭后的反应液升温至40-45℃反应1小时,反应结束后分层除去有机层,将水相以氢氧化钠中和至ph=11~12,然后用四氢呋喃萃取两次,合并的有机相以饱和食盐水洗涤一次后,合并有机相回收溶剂后进行结晶得到5.6g产品,纯度93.5%,收率40%。采用与实施例一相同的方法对所得到的产品进行结构确认,经确认所得到的产品为5-甲氧基-7-氮杂吲哚。实施例七5-甲氧基-7-氮杂吲哚的合成(1)化合物2-氨基-3-甲基-5-甲氧基吡啶的合成将40.0g(0.21mol,1.0eq)的化合物1和200g的甲醇加入到500ml高压釜中,搅拌至固体全部溶解后,依次加入34.7g(0.64mol,3.0eq)的甲醇钠和8.7g(0.11mol,0.5eq)的氧化铜,加热至140-150℃反应8小时,降温至25℃,将釜内物料过滤,滤饼以少量甲醇淋洗,母液浓缩至断流,向其中加入二氯甲烷和水萃取分层,水相用二氯甲烷再萃取一次,合并有机相浓缩至断流,粗品通过40cm填料柱进行减压精馏,收集t=85-90℃/p=2-3mmhg馏分,共计获得12.1g的化合物,纯度98.1%,收率44%;采用与实施例一相同的方法对所得到的化合物进行结构确认,经确认所得到的化合物为2-氨基-3-甲基-5-甲氧基吡啶;(2)化合物2的合成在0℃,向250ml四口瓶加入20.7g(0.15mol,1.0eq)步骤(1)所得到的化合物2-氨基-3-甲基-5-甲氧基吡啶和82.9g乙腈,待物料全部溶解后加入1.1g(0.007mol,0.05eq)4-二甲氨基吡啶,升温至60℃,将32.3g(0.15mol,1eq)二碳酸二叔丁酯溶解在82.9g的乙腈中,并保持四口瓶内温度60-65℃滴加,滴加完毕后在该温度下继续反应8小时得到中间体化合物,然后加入12.0g氢氧化钠(0.30mol,2.0eq),并且在该温度继续反应,反应时间为6小时,反应结束后,以稀盐酸淬灭反应液至中性,升温至80-85℃蒸除溶剂至断流、以四氢呋喃萃取2次,然后合并有机相并蒸干溶剂后进行结晶分出有机层,然后合并有机相并蒸干溶剂后进行结晶,得到25.7g化合物2,纯度95.9%,收率72%;采用与实施例一相同的方法对所得到的化合物2进行结构确认,经确认所得到的化合物2为2-叔丁氧基氨基-3-甲基-5-甲氧基吡啶;(3)产品的合成:向500ml四口瓶通入氮气保护,加入22.6g(0.095mol,1.0eq)步骤(2)所得到的化合物2和121.2g的四氢呋喃,降温至-10±5℃,并控制在该温度下滴加64.6g(0.24mol,2.5eq)2.5m正丁基锂的己烷溶液,滴加完毕,保持-10±5℃搅拌4小时后,在该温度下滴加8.3g(0.11mol,1.2eq)的n,n-二甲基甲酰胺,滴加完毕后,保持-10±5℃搅拌2小时,将瓶中混合物保持-10±5℃慢慢倾倒入68.1g35%的浓盐酸中淬灭反应,全程保持0±5℃淬灭反应,然后将淬灭后的反应液升温至40-45℃反应2小时,除去有机层,将水相以氢氧化钠中和至ph=11-12,然后用甲基叔丁基醚萃取两次,合并的有机相以饱和食盐水洗涤一次后,常压蒸馏除去溶剂得到4.9g产品,纯度93.3%,收率35%。采用与实施例一相同的方法对所得到的产品进行结构确认,经确认所得到的产品为5-甲氧基-7-氮杂吲哚。实施例八5-甲氧基-7-氮杂吲哚的合成(1)化合物2-氨基-3-甲基-5-甲氧基吡啶的合成将40.0g(0.21mol,1.0eq)的化合物1和120g的甲醇加入到200ml高压釜中,搅拌至固体全部溶解后,依次加入12.7g(0.23mol,1.1eq)的甲醇钠和20.0g(0.32mol,1.5eq)的铜粉,加热至100-105℃反应15小时,降温至25℃,将釜内物料过滤,滤饼以少量甲醇淋洗,母液浓缩至断流,向其中加入二氯甲烷和水萃取分层,水相用二氯甲烷再萃取一次,合并有机相浓缩至断流,粗品通过40cm填料柱进行减压精馏,收集t=85-90℃/p=2-3mmhg馏分,共计获得5.6g的化合物,纯度92.5%,收率19%;采用与实施例一相同的方法对所得到的化合物进行结构确认,经确认所得到的化合物为2-氨基-3-甲基-5-甲氧基吡啶;(2)化合物2的合成:在0℃,向2l四口瓶加入20.7g(0.15mol,1.0eq)步骤(1)所得到的化合物2-氨基-3-甲基-5-甲氧基吡啶和414g甲醇,待物料全部溶解后加入0.37g(0.003mol,0.02eq)4-二甲氨基吡啶,升温至60℃,将81.8g(0.38mol,2.5eq)二碳酸二叔丁酯溶解在122.8g的叔丁醇中,并保持四口瓶内温度60-65℃滴加,滴加完毕后保持80-85℃继续反应1小时得到中间体化合物,然后加入30.0g氢氧化钠(0.75mol,5.0eq),并且在该温度继续反应,反应时间为40小时,反应结束后,以稀醋酸淬灭反应液至中性,升温至80-85℃蒸除溶剂至断流、以四氢呋喃萃取2次,然后合并有机相并蒸干溶剂后进行结晶分出有机层,然后合并有机相并蒸干溶剂后进行结晶,得到7.5g化合物2,纯度88.2%,收率21%;采用与实施例一相同的方法对所得到的化合物2进行结构确认,经确认所得到的化合物2为2-叔丁氧基氨基-3-甲基-5-甲氧基吡啶;(3)产品的合成:向3l四口瓶通入氮气保护,加入22.6g(0.096mol,1.0eq)步骤(2)所得到的化合物2和1816g的乙腈,降温至-5℃,并控制在该温度下滴加64.8g(0.24mol,2.5eq)2.5m正丁基锂的己烷溶液,滴加完毕后,保持-5℃继续搅拌6小时,然后在该温度下滴加69.6g(0.96mol,10eq)的n,n-二甲基甲酰胺,滴加完毕后,保持20-25℃搅拌24小时,将瓶中混合物保持-10±5℃慢慢倾倒入45.4g的醋酸中淬灭反应,并保持该温度继续反应6小时后,除去有机层,将水相以氢氧化钠中和至ph=11-12,然后用甲基叔丁基醚萃取两次,合并的有机相以饱和食盐水洗涤一次后,常压蒸馏除去溶剂得到1.6g产品,纯度85.6%,收率11%;采用与实施例一相同的方法对所得到的产品进行结构确认,经确认所得到的产品为5-甲氧基-7-氮杂吲哚。实施例九5-甲氧基7-氮杂吲哚的合成(1)化合物2-氨基-3-甲基-5-甲氧基吡啶的合成将40.0g(0.21mol,1.0eq)的化合物1和20g的甲醇加入到200ml高压釜中,搅拌至固体全部溶解后,依次加入17.0g(0.32mol,1.5eq)的甲醇钠和0.14g(0.01mol,0.05eq)的铜粉,加热至80-85℃反应3小时,降温至25℃,将釜内物料过滤,滤饼以少量甲醇淋洗,母液浓缩至断流,向其中加入二氯甲烷和水萃取分层,水相用二氯甲烷再萃取一次,合并有机相浓缩至断流,粗品通过40cm填料柱进行减压精馏,收集t=85-90℃/p=2-3mmhg馏分,共计获得1.5g的化合物,纯度89.8%,收率5%;采用与实施例一相同的方法对所得到的化合物进行结构确认,经确认所得到的化合物为2-氨基-3-甲基-5-甲氧基吡啶;(2)化合物2的合成在0℃,向1l四口瓶加入20.7g(0.15mol,1.0eq)步骤(1)所得到的化合物2-氨基-3-甲基-5-甲氧基吡啶和103.6g四氢呋喃,然后加入3.7g(0.03mol,0.2eq)4-二甲氨基吡啶,降温至-40℃,保持-40±5℃加入40.8g(0.15mol,1.0eq)2.5m正丁基锂的己烷溶液,并且在该温度继续反应15小时后,将327.4g(1.5mol,10eq)二碳酸二叔丁酯溶解在40.9g的四氢呋喃中,并保持四口瓶内温度-40±5℃滴加,滴加完毕后在该温度下继续反应40小时,反应结束后,以25%盐酸淬灭反应液至中性,升温至80-85℃蒸除溶剂至断流,以四氢呋喃萃取2次,然后合并有机相并蒸干溶剂后进行结晶分出有机层,然后合并有机相并蒸干溶剂后进行结晶,得到13.2g化合物2,纯度89.5%,收率37%;采用与实施例一相同的方法对所得到的化合物2进行结构确认,经确认所得到的化合物2为2-叔丁氧基氨基-3-甲基-5-甲氧基吡啶;(3)产品的合成:向3l四口瓶通入氮气保护,加入22.6g(0.095mol,1.0eq)步骤(2)所得到的化合物2和2270g的二氯甲烷,降温至-5℃,并控制在该温度下滴加51.8g(0.19mol,2.0eq)2.5m正丁基锂的己烷溶液,滴加完毕后保持30±5℃继续搅拌2小时,然后降温至-10±5℃滴加55.7g(0.77mol,8.0eq)的n,n-二甲基甲酰胺,滴加完毕后,保持-10±5℃搅拌6小时,将瓶中混合物缓慢升至10±5℃并保持该温度慢慢倾倒入136.2g30%的柠檬酸中淬灭反应,全程保持10±5℃淬灭反应,继续在该温度下反应6小时,除去有机层,将水相以氢氧化钠中和至ph=11-12,然后用甲基叔丁基醚萃取两次,合并的有机相以饱和食盐水洗涤一次后,常压蒸馏除去溶剂得到2.0g产品,纯度88.7%,收率14%;采用与实施例一相同的方法对所得到的产品进行结构确认,经确认所得到的产品为5-甲氧基-7-氮杂吲哚。实施例十5-甲氧基-7-氮杂吲哚的合成(1)化合物2-氨基-3-甲基-5-甲氧基吡啶的合成将40.0g(0.21mol,1.0eq)的化合物1和2000g的甲醇加入到5l高压釜中,搅拌至固体全部溶解后,依次加入9.3g(0.17mol,0.8eq)的甲醇钠和27.2g(0.43mol,2.0eq)的铜粉,加热至120-125℃反应8小时,降温至25℃,将釜内物料过滤,滤饼以少量甲醇淋洗,母液浓缩至断流,向其中加入二氯甲烷和水萃取分层,水相用二氯甲烷再萃取一次,合并有机相浓缩至断流,粗品通过40cm填料柱进行减压精馏,收集t=85-90℃/p=2-3mmhg馏分,共计获得5.3g的化合物,纯度90.3%,收率18%;采用与实施例一相同的方法对所得到的化合物进行结构确认,经确认所得到的化合物为2-氨基-3-甲基-5-甲氧基吡啶。(2)化合物2的合成在0℃,向2l四口瓶加入20.7g(0.15mol,1.0eq)步骤(1)所得到的化合物2-氨基-3-甲基-5-甲氧基吡啶和621g四氢呋喃,待物料全部溶解后加入18.3g(0.15mol,1.0eq)4-二甲氨基吡啶,降温至0℃;保持该温度滴加82.5g(0.45mol,3.0eq)双三甲基硅基氨基钠后,在该温度下反应6小时;将81.8g(0.38mol,3.0eq)二碳酸二叔丁酯溶解在40.9g的四氢呋喃中,并保持四口瓶内温度0-5℃滴加,滴加完毕后在20-25℃继续反应30小时,反应结束后,以25%盐酸淬灭反应液至中性,升温至80-85℃蒸除溶剂至断流,以四氢呋喃萃取2次,然后合并有机相并蒸干溶剂后进行结晶分出有机层,然后合并有机相并蒸干溶剂后进行结晶,得到18.6g化合物2,纯度95.5%,收率52%;采用与实施例一相同的方法对所得到的化合物2进行结构确认,经确认所得到的化合物2为2-叔丁氧基氨基-3-甲基-5-甲氧基吡啶;(3)产品的合成:向2l四口瓶通入氮气保护,加入22.7g(0.096mol,1.0eq)步骤(2)所得到的化合物2和908g的四氢呋喃,降温至-10±5℃,并控制在该温度下滴加246.9g(0.57mol,6.0eq)2.0m二异丙基氨基锂的己烷溶液,滴加完毕后保持20±5℃继续搅拌4小时,然后降温至-20±5℃并在该温度下滴加34.8g(0.48mol,5.0eq)的n,n-二甲基甲酰胺,滴加完毕后,保持-20±5℃搅拌4小时,将瓶中混合物保持0±5℃慢慢倾倒入113.5g35%的浓盐酸中淬灭反应,全程保持0±5℃淬灭反应,然后将淬灭后的反应液在该温度下继续反应6小时,除去有机层,将水相以氢氧化钠中和至ph=11-12,然后用甲基叔丁基醚萃取两次,合并的有机相以饱和食盐水洗涤一次后,常压蒸馏除去溶剂得到6.4g产品,纯度95.6%,收率45%;采用与实施例一相同的方法对所得到的产品进行结构确认,经确认所得到的产品为5-甲氧基-7-氮杂吲哚。实施例十一5-甲氧基-7-氮杂吲哚的合成(1)化合物2-氨基-3-甲基-5-甲氧基吡啶的合成将40.0g(0.21mol,1.0eq)的化合物1和1200g的乙醇加入到2l高压釜中,搅拌至固体全部溶解后,依次加入231.2g(4.3mol,20.0eq)的甲醇钠和24.5g(0.39mol,1.8eq)的铜粉,加热至150-155℃反应15小时,降温至25℃,将釜内物料过滤,滤饼以少量甲醇淋洗,母液浓缩至断流,向其中加入二氯甲烷和水萃取分层,水相用二氯甲烷再萃取一次,合并有机相浓缩至断流,粗品通过40cm填料柱进行减压精馏,收集t=85-90℃/p=2-3mmhg馏分,共计获得9.2g的化合物,纯度96.6%,收率31%;采用与实施例一相同的方法对所得到的化合物进行结构确认,经确认所得到的化合物为2-氨基-3-甲基-5-甲氧基吡啶;(2)化合物2的合成在0℃,向2l四口瓶加入20.7g(0.15mol,1.0eq)步骤(1)所得到的化合物2-氨基-3-甲基-5-甲氧基吡啶和310.5g四氢呋喃,待物料全部溶解后加入9.2g(0.075mol,0.5eq)4-二甲氨基吡啶,升温至60℃,将36.0g(0.17mol,1.1eq)二碳酸二叔丁酯溶解在1080.3g的四氢呋喃中,并保持四口瓶内温度60-65℃滴加,滴加完毕后在该温度下继续反应15小时得到中间体化合物,然后加入21.0g氢氧化钾(0.38mol,2.5eq),并在20-25℃继续反应,反应时间为8小时,反应结束后,以水淬灭反应液至中性,升温至80-85℃蒸除溶剂至断流,以四氢呋喃萃取2次,然后合并有机相并蒸干溶剂后进行结晶分出有机层,然后合并有机相并蒸干溶剂后进行结晶,得到10.4g化合物2,纯度92.0%,收率29%;采用与实施例一所述的方法对所得到的化合物2进行结构确认,经确认所得到的化合物2为2-叔丁氧基氨基-3-甲基-5-甲氧基吡啶;(3)产品的合成:向2l四口瓶通入氮气保护,加入22.7g(0.096mol,1.0eq)步骤(2)所得到的化合物2和454g的四氢呋喃,降温至-20±5℃,并控制在该温度下滴加64.8g(0.24mol,2.5eq)2.5m正丁基锂的己烷溶液,滴加完毕后保持-20±5℃继续搅拌12小时,然后在该温度下滴加10.4g(0.14mol,1.5eq)的n,n-二甲基甲酰胺,滴加完毕后,保持20±5℃搅拌10小时,将瓶中混合物保持-10±5℃慢慢倾倒入227.0g35%的浓盐酸中淬灭反应,全程保持0±5℃淬灭反应,然后将淬灭后的反应液在该温度下继续反应1小时,除去有机层,将水相以氢氧化钠中和至ph=11-12,然后用甲基叔丁基醚萃取两次,合并的有机相以饱和食盐水洗涤一次后,常压蒸馏除去溶剂得到8.9g产品,纯度97.0%,收率63%;采用与实施例一相同的方法对所得到的产品进行结构确认,经确认所得到的产品为5-甲氧基-7-氮杂吲哚。实施例十二5-甲氧基-7-氮杂吲哚的合成(1)化合物2-氨基-3-甲基-5-甲氧基吡啶的合成将400.0g(2.14mol,1.0eq)的化合物1和2kg的甲醇加入到5l高压釜中,搅拌至固体全部溶解后,依次加入346.6g(6.42mol,3.0eq)的甲醇钠和68.0g(1.07mol,0.5eq)的铜粉,加热至130℃反应8小时,降温至25℃,将釜内物料过滤,滤饼以少量甲醇淋洗,母液浓缩至断流,向其中加入二氯甲烷和水萃取分层,水相以二氯甲烷再萃取一次,合并有机相浓缩回收溶剂,粗品通过40cm填料柱进行减压精馏,收集t=85-90℃/p=2-3mmhg馏分,共计获得153.5g的化合物,纯度98.4%,收率52%;采用与实施例一相同的方法对所得到的化合物进行结构确认,经确认所得到的化合物为2-氨基-3-甲基-5-甲氧基吡啶;(2)产品的合成向2l四口瓶加入48.0g(0.35mol,1.0eq)步骤(1)所得到的化合物2-氨基-3-甲基-5-甲氧基吡啶和240g四氢呋喃,待物料全部溶解后于-5℃向反应液中滴加207.9g(0.76mol,2.2eq)2.5m的正丁基锂的己烷溶液,-5℃反应1-2小时后,然后将83.4g(0.38mol,1.1eq)二碳酸二叔丁酯于-5℃加入到反应液中,并保持-5℃反应3小时,然后于-5℃向反应液中滴加207.9g(0.76mol,2.2eq)2.5m的正丁基锂的己烷溶液,并保持-5℃反应1-2小时,保持-5℃将30.5g(0.42mol,1.2eq)n,n-二甲基甲酰胺滴加到上述反应液中,并继续再该温度下反应6-8小时,将反应混合物用盐酸淬灭,分层,水层以碱中和至ph=11-12,用有机溶剂萃取水层,水洗有机层,有机层经回收溶剂后,结晶得到7.0g的产品,纯度96.6%,收率14%;采用与实施例一相同的方法对所得到的产品进行结构确认,经确认所得到的产品为5-甲氧基-7-氮杂吲哚。综上所述,本发明所述的制备方法,与目前报道的合成方法相比大大缩短了合成路线,本发明所述的原料2-氨基-3-甲基-5-溴嘧啶,与现有工艺的5-溴-7-氮杂吲哚相比,原料廉价易得,有效地避免了现有技术的产物,所得产品的纯度相对较高;并且本发明所用的原料以及试剂均为廉价易得,非常易于实现工业化。以上所述,仅是本发明实施的较佳实施例,并非对本发明做任何形式上的限制,凡在本发明的精神和原则之内所做的修改、等同替换和改进等,均需要包含在本发明的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1