具有低多分散指数(PDI)的聚丙烯腈(PAN)聚合物及由其制成的碳纤维的制作方法
本公开内容总体涉及聚丙烯腈(pan)聚合物的合成以及由pan聚合物形成碳纤维的方法。
背景技术:
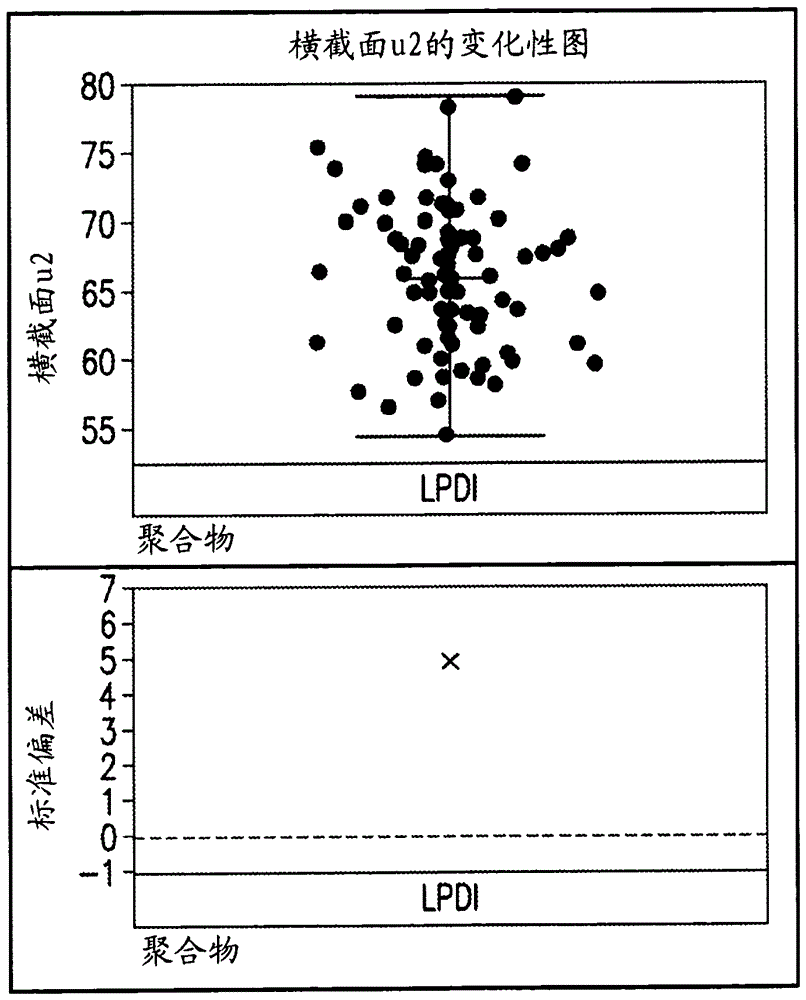
:由于诸如高比强度和比刚度、高耐化学性和低热膨胀的性质,碳纤维已经广泛用于航空航天、体育运动,和汽车、风能以及其它节能领域的商业化行业。通常,碳纤维由基于聚丙烯腈(pan)的聚合物制成。自由基聚合传统上,pan聚合物通过自由基聚合方法制成。在自由基聚合中,催化剂或引发剂首先引发以形成起始自由基物质。这些自由基物质开始与单体反应以产生活性中心,从而形成游离的单体自由基。然后,所述单体自由基与其它单体反应以增长分子链,从而形成聚合物自由基。在聚合期间,作为偶合终止,有时一个自由基与另一个自由基反应以偶合并形成长死链,而一条链末端上的一些自由基可以攻击第二自由基链中的第二个至最后一个碳原子上的氢原子以形成歧化终止。聚合物自由基还可以与另一种化合物如链转移剂反应,以终止聚合物自由基的增长反应并且由链转移剂形成新的自由基。该新形成的链转移自由基开始其新的链增长。因此,链转移剂减小了增长的聚合物自由基链的长度。如果该终止的速率比增长的速率高得多,则形成具有短链长度的非常小的聚合物。因此,链转移剂用于控制聚合物的分子长度或分子量。由于不同的终止机制,所得分子链具有不同长度或不同分子量。因此,聚合物的分子量具有一定分布。该分布可通过其多分散指数来定义,如下:或者,pdi可以表示如下:。mw、mn、mz通过gpc(凝胶渗透色谱)方法来测量。在此,mw是重均分子量。mn是数均分子量并且mz是z-均分子量或尺寸平均分子量。高pdi表明聚合物具有大的分子量分布,这意味着聚合物具有非常高的分子量物质或非常低的分子量物质,或两者兼有。换句话说,所述聚合物由长度变化很大的分子链组成。过高分子量或过小分子量物质的存在将影响聚合物通过纺丝成为纤维的加工性以及所得纤维的性质,尤其是过小分子量物质,由于小分子量物质是聚合物机械性质的一类分子缺陷的事实。通过常规自由基聚合制备的pan聚合物不允许在聚合上控制。所得聚合物具有大的分子量分布。因此,由这样的pan纺成的纤维的机械性质发展存在困难。发明概述本公开内容提供一种用于合成具有窄分子量分布的聚丙烯腈(pan)聚合物的方法,以及由这样的聚合物制造碳纤维前体的方法。优选的pan聚合物具有约2或更小的pdi(mw/mn)。使用特定raft(可逆加成-断裂链转移)剂,通过可控/活性自由基聚合来合成这样的pan聚合物。由纤维前体制造的碳纤维呈现出良好性质,如均匀横截面、低微观缺陷和分子缺陷。这样的良好特性是由于低pdi聚合物具有均匀mw并在碳纤维生产工艺期间导致低分子缺陷和微观缺陷的事实。附图简要说明图1是显示由低pdi的pan聚合物制造的冷冻干燥的pan凝结纤维的微孔分布的水银孔率法曲线图。图2是由低pdi的pan聚合物制造的pan纤维前体的横截面区域的显微图像。图3是图2中提及的相同pan纤维前体的横截面区域的变化性图。发明详述本公开内容的一方面涉及一种使用特定raft剂通过可控/活性自由基聚合来控制pan分子量分布或pdi的机制。pdi(mw/mn)目标为约2或更小,优选1.2-1.9的pdi(mw/mn)(或替代的1.2-1.7的pdi(mz/mw)。可控/活性自由基聚合如果在自由基聚合期间链终止仅发生在所有单体被消耗之后,则该聚合称为活性聚合。在这种聚合反应中,如果将更多的单体添加到反应中,则增长可继续进行。作为理想的活性聚合,所有链在反应开始时被引发并以相似速率增长。没有不可逆的链转移或终止。如果引发相对于增长是快速的,则分子量分布非常窄并且可通过进一步向反应中添加单体来延长所述链。然而,在自由基聚合中,所有链无法同时是活性的。因此,使用一些试剂通过形成休眠阶段来控制增长及其速率。通过可逆地钝化或活化增长,可实现活性链与休眠链之间的快速平衡,以在相似的速率下控制链增长,使得可获得窄分子量分布。这称为“可控/活性自由基聚合”。本文使用的化学品称为raft(可逆加成/断裂链转移)剂。pan聚合物的合成制备具有窄分子量分布的pan聚合物的方法是溶液聚合法,其包括:a.使丙烯腈(an)单体与溶剂、一种或多种共聚单体,以及raft剂(如本文所定义的)结合以形成溶液;b)将所述溶液加热至高于室温的温度,即>25℃,例如40℃-85℃;和c)将引发剂添加至溶液,以引发聚合反应。在聚合完成之后,除去未反应的an单体,例如在高真空下脱气,并冷却所得pan聚合物溶液。在此阶段,pan聚合物为准备用于纺丝的溶液或纺丝原液形式。使用raft剂,通过可控/活性自由基聚合来影响an单体的聚合,所述raft剂为具有以下结构的硫代羰基硫代化合物:所述raft剂的有效性取决于取代基r和z。所述取代基影响聚合反应动力学和结构控制的程度。r基团是自由基离去基团。其控制raft聚合期间的再引发聚合。并且z基团控制c=s键反应性的稳定性并影响自由基加成和断裂的速率。优选的raft剂选自具有以下结构的硫代羰基硫代化合物:(ⅰ)其中z1=-ch2-(ch2)10-ch3;-(ch2)n-ch3,n=0-20; -c(ch3)m-cooh,m=1-2;-c(ch3)m-cooch3,m=1-2; -c(ch3)m-cooc2h5,m=1-2;r1=r’1=-cn;,x=0-1;r”1=h;-ch3;-(ch2)m-cooh,m=1-2;r”’1=h;-ch3(ⅱ)其中z2=r=f、cl、cn、och3;r2=r’2=-cn;,x=0-1; -c(ch3)m-cooch3,m=1-2; -c(ch3)m-cooc2h5,m=1-2;r”2=h;-ch3;-(ch2)m-cooh,m=1-2;r”’2=h;-ch3(ⅲ)其中z3=-ch2-(ch2)10-ch3;-(ch2)y-ch3,y=1-20;r3=-ch2-(ch2)10-ch3;-(ch2)y-ch3,y=1-20;具有上述结构ⅰ、ⅱ和ⅲ的raft剂的具体实例分别为:1)三硫代碳酸酯:2-氰基-2-丙基十二烷基三硫代碳酸酯(cpdtc)2)二硫代苯甲酸酯:2-氰基-2-丙基苯并二硫代酸酯(2-cyano-2-propylbenzodithioate)(cpbz)3)硫代羰基二硫化物:双-十二烷基硫烷基硫代羰基二硫化物(bdstd)。用于聚合的合适的溶剂包括:二甲基亚砜(dmso)、二甲基甲酰胺(dmf)、二甲基乙酰胺(dmac)、碳酸亚乙酯(ec)、氯化锌(zncl2)/水,以及硫氰酸钠(nascn)/水。适用于pan聚合物合成的共聚单体可以是一种或多种基于乙烯基的酸,包括:甲基丙烯酸(maa)、丙烯酸(aa)、衣康酸(ita);基于乙烯基的酯,例如甲基丙烯酸酯(ma)、甲基丙烯酸甲酯(mma)、乙酸乙烯酯(va)、丙烯酸乙酯(ea)、丙烯酸丁酯(ba)、甲基丙烯酸乙酯(ema),以及其它乙烯基衍生物,例如乙烯基咪唑(vim)、丙烯酰胺(aam)和双丙酮丙烯酰胺(daam)。pan聚合可通过以下引发剂(或催化剂)来引发:基于偶氮的化合物,例如:偶氮二异丁腈(aibn)、偶氮二氰基戊酸(acva)和2,2’-偶氮双-(2,4-二甲基)戊腈(abvn),或其它,或有机过氧化物,例如,过氧化二月桂酰(lpo)、二叔丁基过氧化物(tbpo)、过氧化二碳酸二异丙酯(ipp)及其它。根据优选的实施方案,基于以下制剂以及足够量的溶剂进行pan聚合以形成含有5wt%-28wt%,优选15wt%-25wt%的最终pan聚合物的溶液,所述制剂:重量%(wt%):>90%an单体;<5%共聚单体;<1%引发剂;<1%raft剂,基于这四种组分的总重量计。可控/活性自由基聚合方法能够控制聚合物构造。这包括分子量、分子量分布(即多分散性)、官能度,以及组成。上述raft剂在将an单体可控/活性自由基聚合成pan期间起链转移剂的作用。raft聚合机制具有四个反应步骤:引发、加成-断裂、再引发和平衡,如下所示,使用例如cpdtc作为raft剂。在pan聚合期间,偶氮二异丁腈(aibn)用作引发剂,并且dmso用作溶剂。a.通过aibn(偶氮二异丁腈)引发。b.使用cpdtc加成-断裂c.再引发。d.平衡通过aibn引发聚合。其分解以形成两个自由基(反应式1),然后所述自由基开始与an单体反应以引发聚合(反应式2)。更多(an)与自由基反应并形成活性聚合物或聚合自由基pn·(反应式3)。cpdtc作为raft剂反应或添加至pn·以形成raft加合物自由基。该raft加合物自由基可导致在任一方向上的断裂反应以得到起始物质或新的自由基和聚合的raft-pn(反应式4)。这是可逆的步骤。在反应式5中,新形成的自由基再引发聚合物增长以得到另一活性聚合物或聚合自由基pm·。该活性聚合物pm·与聚合的raft-pn反应以形成raft加合物自由基中间体(反应式6)。该中间体可在任一方向上断裂以控制对于pn·或pm·增长具有同等机会和窄pdi的链。当所有单体和共聚单体被耗尽时,聚合将结束。通过上述方法制造的pan聚合物的分子量可以在60-500千克/摩尔,优选90-250千克/摩尔,以及最优选115-180千克/摩尔的范围内,其中pdi为约2或更小。通过viscotekgpcmax凝胶渗透色谱(gpc)系统测量分子量。在表征期间,具有0.02mlibr的dmf(二甲基甲酰胺)用作具有1ml/分钟流量的流动相。并且柱温度设定为45℃。碳纤维制造上述低pdi的pan聚合物适合用于湿纺或气隙纺丝(或替代地“干喷湿纺”)以制成连续的碳纤维前体(即白色纤维)。已经发现,低pdi的pan聚合物具有良好的纺丝能力;即易于通过纺丝工艺由这样的聚合物制成纤维。根据astm2256,由这样的聚合物制造的所得纤维前体显示出横截面均匀性、韧性>5克/旦,和初始模量>125克/旦。为制作pan白色纤维,在通过真空除去气泡后,使pan聚合物溶液(即纺丝“原液”)经历常规湿纺和/或气隙纺丝。该纺丝“原液”可具有5wt%-28wt%,优选15wt%-25wt%范围内的聚合物浓度,基于溶液的总重量计。在湿纺中,将原液过滤并经喷丝头(由金属制成)的孔挤出到用于聚合物的液体凝结浴中以形成长丝。喷丝头孔决定pan纤维的期望长丝数(例如,对于3k碳纤维3000个孔)。在气隙纺丝中,在喷丝头和凝结浴之间提供1-50mm,优选2-15mm的垂直气隙。在这种纺丝法中,将聚合物溶液过滤并在空气中从喷丝头挤出,然后将挤出的长丝在凝结浴中凝结。该方法中使用的凝结液体是溶剂和非溶剂的混合物。水或醇通常用作非溶剂。溶剂与非溶剂的比率以及浴温度用于调整挤出的初生长丝在凝结中的固化速率。然后,通过辊将纺成的长丝从凝结浴取出,通过洗涤浴以除去过量的凝结剂并在热水浴(例如40℃-100℃)中拉伸以赋予长丝分子取向,作为控制纤维直径的第一步骤。然后在例如干燥辊上将拉伸的长丝干燥。干燥辊可以由多个串联并且以蛇形配置布置的可旋转辊构成,在其上长丝从辊到辊顺序通过并且在足够的张力下以提供纤维在辊上的拉伸或松弛。借助于在内部或通过辊循环的加压蒸汽或在辊的内部的电加热元件加热至少一些辊。在干燥之前,可以将纺丝油剂施加至经拉伸的纤维上,以防止长丝在下游工艺中彼此粘附。作为控制纤维直径的第二步骤,在第一纤维拉长之后进行超级拉伸。该超级拉伸工艺在100℃-185℃(高于纤维的玻璃化转变温度),优选135℃-175℃的温度下进行。这样的拉伸进一步将分子定向为长丝。经超级拉伸的纤维可具有约0.4-1.5旦、优选0.5-1.0旦的直径。使处理条件(包括纺丝溶液和凝结浴的组成、总拉伸的量、温度,和长丝速度)相关联以提供期望结构和旦的长丝。在超级拉伸步骤之后,纤维长丝可通过一个或多个热辊,然后缠绕到筒管上。为将pan白色纤维转化为碳纤维,使pan纤维经历氧化和碳化。在氧化阶段期间,将pan纤维在张力下进料通过一个或多个专门的烘箱,将热空气进到所述专门的烘箱中。氧化烘箱温度可在200℃-300℃,优选220℃-285℃范围内。氧化工艺使来自空气的氧分子与pan纤维结合并导致聚合物链开始交联,从而使纤维密度增加至1.3g/cm3-1.4g/cm3。在氧化工艺中,施加至纤维的张力通常是控制拉长或收缩的纤维在0.8-1.35、优选1.0-1.2的拉伸比下。当拉伸比是1时,没有拉伸。当拉伸比大于1时,施加的张力导致纤维被拉伸。这样氧化的pan纤维具有不熔性阶梯芳族分子结构,并且其准备用于碳化处理。碳化发生在一个或多个专门设计的炉内部的惰性(无氧)气氛中。在优选的实施方案中,氧化的纤维穿过预碳化炉,使纤维经历约300℃-900℃、优选350℃-750℃的加热温度,同时暴露于惰性气体,例如氮气,接着通过使纤维穿过加热至约700℃-1650℃、优选800℃-1450℃的更高温度的炉来碳化,同时暴露于惰性气体。应在整个预碳化和碳化工艺中添加纤维张力。在预碳化中,施加的纤维张力足以将拉伸比控制在0.9-1.2、优选1.0-1.15的范围内。在碳化中,所用的张力足以提供0.9-1.05的拉伸比。碳化导致碳分子的结晶并由此制造具有大于90%碳含量的最终碳纤维。基质树脂与碳纤维之间的粘合力是碳纤维增强的聚合物复合材料中的重要标准。因此,在碳纤维制造期间,可在氧化和碳化之后进行表面处理以增强该粘合力。表面处理可包括牵引碳化纤维通过包含电解质如碳酸氢铵或次氯酸钠的电解浴。电解浴的化学品蚀刻或粗糙化纤维表面,从而增加可用于界面纤维/基质粘合的表面积,并添加反应性化学基团。接着,可使碳纤维经历上浆,其中将上浆涂料如基于环氧树脂的涂料施加到所述纤维上。通过使纤维穿过含有液体涂布材料的上浆浴来进行上浆。在处理和加工成中间体形式(如干织物和预浸料)期间,上浆保护碳纤维。上浆还将长丝一起保持在单个丝束中以减少细毛,改进加工性并增加纤维和基质树脂之间的界面剪切强度。在上浆之后,将经涂布的碳纤维干燥并缠绕到筒管上。已经发现,由上述低pdi的pan聚合物制造的碳纤维具有以下机械性质:根据astmd4018测试方法,拉伸强度大于700ksi(4826mpa),以及拉伸初始模量大于35msi(241gpa)。将通过以下实施例进一步阐明上述pan聚合物和由其制造的碳纤维的益处及性质。实施例实施例1pan聚合物的合成根据表1a-1c所示的用于pan聚合的制剂来制备pan聚合物。表1a—用于pan聚合的制剂组分制剂1制剂2制剂3制剂4丙烯腈(an)99.3099.3099.3099.30衣康酸(ita)0.700.700.700.70cpbz0.113%0.029%bdstd0.359%*0.045%表1b—用于pan聚合的制剂组分制剂5制剂6制剂7制剂8丙烯腈(an)99.3099.3099.0098.00衣康酸(ita)0.700.701.00甲基丙烯酸(maa)2.00cpdtc0.019%0.009%0.022%0.033%表1c—用于pan聚合的制剂组分制剂9制剂10制剂11制剂12丙烯腈(an)96.0096.0097.0099.00衣康酸(ita)1.001.00甲基丙烯酸(maa)2.002.00甲基丙烯酸酯(ma)2.002.00乙烯基咪唑(vim)2.00cpdtc0.030%0.030%0.025%0.022%在上表中,cpdtc、cpbz、bdstd为raft剂,其中:cpdtc=2-氰基-2-丙基十二烷基三硫代碳酸酯cpbz=2-氰基-2-丙基苯并二硫代酸酯bdstd=双-十二烷基硫烷基硫代羰基二硫化物注意:*以摩尔%使用raft剂,基于单体总量计。如下进行可控/活性自由基pan聚合。偶氮二异丁腈(aibn)用作引发剂/催化剂并且dmso用作溶剂。raft剂用作链转移剂。在聚合期间,进行以下顺序的步骤:a)将dmso从dmso储存罐计量至反应器,然后将an从an储存罐计量至反应器;b)用氮气吹扫所述反应器;c)预加热所述反应器并且在高于室温(25℃)下,将共聚单体和raft剂添加至反应器中;d)加热所述反应器,然后在40-85℃的期望温度点下添加引发剂/催化剂;e)在60-80℃的温度下开始聚合,进行15-23小时的时间;f)冷却至低温(40-50℃)并排出聚合物溶液。在聚合之后,测量制造的pan聚合物的分子量和pdi,并且结果显示于表2a-2c中。使用凝胶渗透色谱(gpc)分析所得pan聚合物的分子量和多分散指数(pdi)。使用具有低角度和直角光散射检测器和ri检测器的viscotekgpcmax/sec色谱系统。收集数据并使用viscotekomnisec4.06版软件分析以确定绝对重均分子量(mw)及其分布。表2a--聚合物分子量及分布制剂1制剂2制剂3制剂4mn(克/摩尔)37101406175677748177mw(克/摩尔)591796336210114382538mw/mn1.5951.5601.7811.713mz8174782742148522120826mz/mw1.3811.3061.4681.464表2b--聚合物分子量及分布制剂5制剂6制剂7制剂8mn(克/摩尔)789451287738612564265mw(克/摩尔)155568217778159746113551mw/mn1.9711.6911.8551.767mz236895327687226813167536mz/mw1.5231.5051.4201.475表2c--聚合物分子量及分布制剂9制剂10制剂11制剂12mn(克/摩尔)72193667106956076579mw(克/摩尔)147459121290137019150027mw/mn2.0431.8181.9701.959mz237764173392195056224951mz/mw1.6121.4301.4241.499由具有raft剂的制剂制造的所有pan聚合物产出具有约2或更小pdi(mw/mn)的pan聚合物。在相对于制剂5调整raft剂的剂量和溶液浓度后,由制剂6制造的pan聚合物具有217778克/摩尔的更高分子量(mw)及1.69的pdi。实施例2白色纤维的制造使用如实施例1所描述的由制剂5制造的pan聚合物通过湿纺形成碳纤维前体(或白色纤维)。使用如实施例1所描述的由制剂12制造的pan聚合物在150μm喷丝头的情况下通过气隙纺丝方法形成白色纤维。如下测定白色纤维的性质。横截面分析将白色纤维束样品浸入到丙烯酸树脂中,然后固化。使用不同等级的砂光机纸在滚球(grounder)上将固化的纤维树脂棒抛光,以获得光滑横截面。之后,在具有图像分析系统的光学显微镜下测量纤维横截面的横截面均匀性。孔隙度测定法对于气隙纺丝,将离开凝结浴的纤维样品在-60℃下冷冻干燥,并且通过水银孔率计测试所述冷冻干燥的样品进行孔隙度和多孔结构分析。韧性&模量根据astmd2256方法测量纤维韧性和初始模量。表3—白色纤维性质&纺丝方法制剂制剂5制剂12原液浓度%18.822.14纺丝方法湿纺气隙喷丝头尺寸3k3k冷冻干燥的凝结纤维孔隙度/%--85.75总拉长比/时间12.210.67白色纤维韧性克/旦7.006.54白色纤维模量克/旦144.3161发现基于制剂5和12的pan聚合物具有良好的纺丝能力。如从表3可看到的,来自湿纺和气隙纺丝两者的所得白色纤维前体还具有良好的韧性和模量。图1是冷冻干燥的凝结纤维中孔隙直径分布的水银孔率法曲线图。y轴是以ml/g或dv/dlogd的log微分压入量。v是压入到样品的孔隙中的水银的体积。x轴是孔隙直径对数。因此,所述图显示了压入体积相对于孔隙直径对数的导数。总体积或空隙为曲线下的面积。图1显示了由根据制剂12的低pdi的pan聚合物通过气隙纺丝制造的冷冻干燥的pan凝结纤维具有低微孔缺陷。图2的显微图像和图3的变化性图显示了通过气隙纺丝纺成的低pdi的白色纤维具有均匀的横截面。图3是横截面面积的变化性图,其显示出分散或扩散。将白色纤维转化为碳纤维在空气中,在220℃-285℃的温度范围内将白色纤维前体氧化,并在氮气中,在350℃-650℃(预碳化)和然后800℃-1300℃的温度范围内碳化。测定所得碳纤维的拉伸强度和拉伸模量并显示于表4中。表4—碳化&碳纤维性质制剂制剂5制剂12氧化温度(℃)220-285220-285预碳化温度(℃)350-650350-650碳化温度(℃)800-1300800-1300纤维拉伸强度(ksi)772(5323mpa)800(5516mpa)纤维拉伸模量(msi)41.9(289gpa)43.0(296gpa)纤维密度(g/cm3)1.8091.822根据astmd4018测定碳纤维的拉伸强度和初始模量。首先将碳纤维浸渍到环氧树脂浴中,然后固化。以0.5英寸/分钟的十字头速度在mts上测试经固化的碳纤维股(strand)的拉伸强度和模量。根据astmd3800,通过液体浸渍法测定纤维密度。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1