一种艾瑞昔布中间体及其制备方法和用途与流程
本发明属于化学医药领域,具体涉及一种艾瑞昔布中间体及其制备方法和用途。
背景技术:
艾瑞昔布(imrecoxib),化学名为1-正丙基-3-(4-甲基苯基)-4-(4-甲磺酰基苯基)-2,5-二氢-1h-2-吡咯酮,化学结构式如式y所示。艾瑞昔布为非甾体抗炎药,用于缓解骨关节炎的疼痛症状,目前已经获得国家药监局批准上市。由于艾瑞昔布缓解疼痛的效果良好,受到研究着广发重视。目前,已经有多个专利报道艾瑞昔布的合成。
专利cn108707100a公开了一种艾瑞昔布及其中间的制备方法,在改制备方法中,采用α-氨基酮在碱性条件下反应,制备得到艾瑞昔布中间体。但是,由《strategicapplicationsofnamedreactioninorganicsynthesis:backgroundanddetailedmechanisms》(lászlókürtiandbarbaraczakó)可知:α-氨基酮在碱性条件下不稳定,容易发生自聚反应。若采用稳定更好的α-氨基酮的盐酸盐制备艾瑞昔布中间体,因为后续步骤中会用到较强的碱,会使反应较为复杂,不利于工业生产。
专利cn1134413c、us20040029951、cn102206178b和cn104193664b等,制备艾瑞昔布中间体,及利用该中间体制备艾瑞昔布时,存在收率低、产品不易分离纯化、试剂昂贵、试剂及反应产生的“三废”毒性大、环境不友好等问题。
技术实现要素:
为了解决上述问题,本发明提供了一种式ⅰ所示的化合物、或其药学上可接受的盐、或其立体异构体、或其溶剂合物、或其前体药物、或其代谢产物:
其中,r选自氢、取代或未取代的c1~c8烷基;所述取代基为卤素、羟基、氰基、硝基、氨基。
进一步地,所述化合物如式ⅰ-a所示:
本发明还提供了一种式ⅱ所示的化合物、或其药学上可接受的盐、或其立体异构体、或其溶剂合物、或其前体药物、或其代谢产物:
其中,r选自氢、取代或未取代的c1~c8烷基;所述取代基为卤素、羟基、氰基、硝基、氨基。
进一步地,所述化合物如式ⅱ-a所示:
本发明还提供了一种前述的化合物的制备方法,它包括如下步骤:
式ⅲ所示化合物在碱的参与下,以有机溶剂为溶剂,与丙酰氯反应,得式ⅰ所示化合物;
其中,r选自氢、取代或未取代的c1~c8烷基;所述取代基为卤素、羟基、氰基、硝基、氨基。
进一步地,它包括如下步骤:
化合物4在碱的参与下,以有机溶剂为溶剂,与丙酰氯反应,得化合物5。
进一步地,所述的碱为三乙胺;所述的有机溶剂为乙腈;化合物4、丙酰氯与三乙胺的摩尔比为35:32~52:42~62;化合物4与乙腈的质量体积比为1:5~25(w/v)。
进一步地,化合物4、丙酰氯与三乙胺的摩尔比为35:42:52;化合物4与乙腈的质量体积比为1:10(w/v)。
进一步地,所述的反应为在室温下反应1~3h,优选为在室温下反应1~2h。
进一步地,与丙酰氯反应后还包括以下步骤:将反应液淬灭、提纯;优选地,所述的淬灭为向反应液中加水淬灭反应;所述的提纯为旋去乙腈和过量的三乙胺,萃取,洗涤并干燥有机相。
进一步地,所述提纯为旋去乙腈和过量的三乙胺,再用乙酸乙酯萃取,饱和食盐水洗涤有机相,无水硫酸钠干燥有机相,并旋干有机相。
进一步地,所述式ⅲ所示化合物的制备方法包括如下步骤:
步骤1:化合物1和nh2oh·hcl于碱的水溶液中反应得反应液,将反应液提纯后得化合物2;
步骤2:化合物2、磺酰氯类化合物和碱于有机溶剂中反应得反应液,将反应液提纯后得化合物3;
步骤3:化合物3和醇钾于无水有机溶剂中反应得反应液,在反应液中加有机溶剂,析出固体,固液分离,向滤液中通入气态酸或酸的无水有机溶液,再提纯,即得式ⅲ所示化合;
其中,r选自氢、取代或未取代的c1~c8烷基;所述取代基为卤素、羟基、氰基、硝基、氨基。
进一步地,当所述式ⅲ所示化合物为化合物4时,所述化合物4的制备方法包括如下步骤:
步骤1:化合物1和nh2oh·hcl于碱的水溶液中反应得反应液,将反应液提纯后得化合物2;
步骤2:化合物2、磺酰氯类化合物和碱于有机溶剂中反应得反应液,将反应液提纯后得化合物3;
步骤3:化合物3和醇钾于无水有机溶剂中反应得反应液,在反应液中加有机溶剂,析出固体,固液分离,向滤液中通入气态酸或酸的无水有机溶液,再提纯,即得化合物4。
进一步地,
步骤1中,所述nh2oh·hcl为nh2oh·hcl水溶液;所述化合物1与nh2oh·hcl水溶液的质量体积比为1~5:1w/v;所述化合物1与碱的水溶液的质量体积比为1:1~5w/v;
和/或,步骤2中,所述化合物2、磺酰氯类化合物、碱的质量比为1:0.5~2.5:0.5~2.5;所述化合物2与有机溶剂的质量体积比为1:10~30w/v;
和/或,步骤3中,所述化合物3与醇钾的质量比为1:0.1~0.5;所述化合物3与无水有机溶剂的质量体积比为1:10~20w/v;所述化合物3与有机溶剂的质量体积比为1:10~20w/v。
进一步地,
步骤1中,所述化合物1与nh2oh·hcl水溶液的质量体积比为2:1w/v;所述化合物1与碱的水溶液的质量体积比为1:3w/v;
和/或,步骤2中,所述化合物2、磺酰氯类化合物、碱的质量比为1:1.35:1.25;所述化合物2与有机溶剂的质量体积比为1:20w/v;
和/或,步骤3中,所述化合物3与醇钾的质量比为1:0.23;所述化合物3与无水有机溶剂的质量体积比为1:15w/v;所述化合物3与有机溶剂的质量体积比为1:15w/v。
进一步地,
步骤1中,所述碱的水溶液为naoh,koh,csoh或k2co3的水溶液;所述碱的水溶液的浓度为10~30%w/v;所述nh2oh·hcl水溶液的浓度为70~90%w/v;
和/或,步骤2中,所述磺酰氯类化合物为对甲基苯磺酰氯;所述碱为有机碱或无机碱;所述有机溶剂为二氯甲烷;
和/或,步骤3中,所述醇钾为甲醇钾;所述无水有机溶剂为无水乙醇;所述有机溶剂为甲基叔丁基醚;所述气态酸或酸的无水有机溶液中酸为盐酸。
进一步地,
步骤1中,所述碱的水溶液为naoh的水溶液;所述碱的水溶液的浓度为20%w/v;所述nh2oh·hcl水溶液的浓度为84%w/v;
和/或,步骤2中,所述碱为氢氧化钾。
进一步地,
步骤1中,所述反应为在冰浴反应1~4h;所述提纯为乙酸乙酯萃取反应液后,合并有机相,干燥;
和/或,步骤2中,所述反应为在室温下反应3~6h;所述提纯为将反应液抽滤、洗涤后干燥;
和/或,步骤3中,所述反应为在60~80℃反应3~6h;所述通入气态酸或酸的无水有机溶液至滤液ph为2~4;所述提纯为将滤液干燥,加入乙酸乙酯后,调节ph至9~11,萃取,干燥有机相,并酸碱倒置。
进一步地,
步骤1中,所述反应为在冰浴反应2h;所述提纯为乙酸乙酯萃取反应液后,合并有机相,有机相用无水naso4干燥,再旋干;
和/或,步骤2中,所述反应为在室温下反应4~5h;所述提纯为将反应液抽滤、洗涤后旋干;
和/或,步骤3中,所述反应为在70℃反应4~5h;所述通入气态酸或酸的无水有机溶液至滤液ph为3;所述提纯为将滤液旋干,加入乙酸乙酯后,naoh调节ph至10,萃取,旋干有机相,并酸碱倒置。
本发明还提供了一种前述的化合物的制备方法,它包括如下步骤:
在氮气保护下,对前述的化合物进行还原,得式ⅱ所示化合物;
其中,r选自氢、取代或未取代的c1~c8烷基;所述取代基为卤素、羟基、氰基、硝基、氨基。
进一步地,它包括如下步骤:
在氮气保护下,对前述的化合物进行还原,得化合物6。
进一步地,所述的还原为前述的化合物与还原剂在有机溶剂中还原;优选地,所述的还原剂为硼烷四氢呋喃溶液;所述的有机溶剂为四氢呋喃;前述的化合物与硼烷四氢呋喃溶液的质量体积比为1:1~10(w/v);前述的化合物与四氢呋喃的质量体积比为1:1~5(w/v);其中,所述烷四氢呋喃溶液的浓度为1~5mol/l。
进一步地,前述的化合物与硼烷四氢呋喃溶液的质量体积比为1:5.8(w/v);前述的化合物与四氢呋喃的质量体积比为1:2(w/v);其中,所述烷四氢呋喃溶液的浓度为1mol/l。
进一步地,所述的还原为在60~100℃下回流反应1~3h;优选在80℃下回流反应1h。
进一步地,对前述的化合物进行还原后还包括以下步骤:将反应液淬灭、提纯;优选地,所述的淬灭为向反应液中加meoh淬灭反应;所述的提纯为旋干反应液,加入稀盐酸后洗涤,并调节水相ph至9~11,萃取,洗涤并干燥有机相。
进一步地,所述提纯为旋干反应液,加入浓度为1mol/l的稀盐酸,用乙酸乙酯洗涤3次,水相用20%的naoh溶液调节ph至10,乙酸乙酯萃取3次,饱和食盐水洗涤有机相,无水硫酸钠干燥有机相,并旋干有机相。
本发明还提供了前述的化合物、或其药学上可接受的盐、或其立体异构体、或其溶剂合物、或其前体药物、或其代谢产物在作为中间体制备艾瑞昔布中的应用。
本发明中w/v为质量体积比,单位为g/ml。
本发明中v/w为体积质量比,单位为ml/g。
本发明中碳氢基团中碳原子含量的最小值和最大值通过前缀表示,例如,前缀(ca~cb)烷基表明任何含“a”至“b”个碳原子的烷基。因此,例如,c1~c8烷基是指包含1~8个碳原子的直链或支链烷基。
本发明中“取代”是指分子中的氢原子被其它不同的原子或分子所替换。
本发明提供了一种艾瑞昔布中间体及其制备方法。本发明艾瑞昔布中间体十分稳定;且制备该艾瑞昔布中间体和利用该艾瑞昔布中间体制备艾瑞昔布的过程中,反应条件温和,试剂毒性小,对环境友好;此外,反应过程中产物易分离纯化,且产率高,适合大规模的工业生产。
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
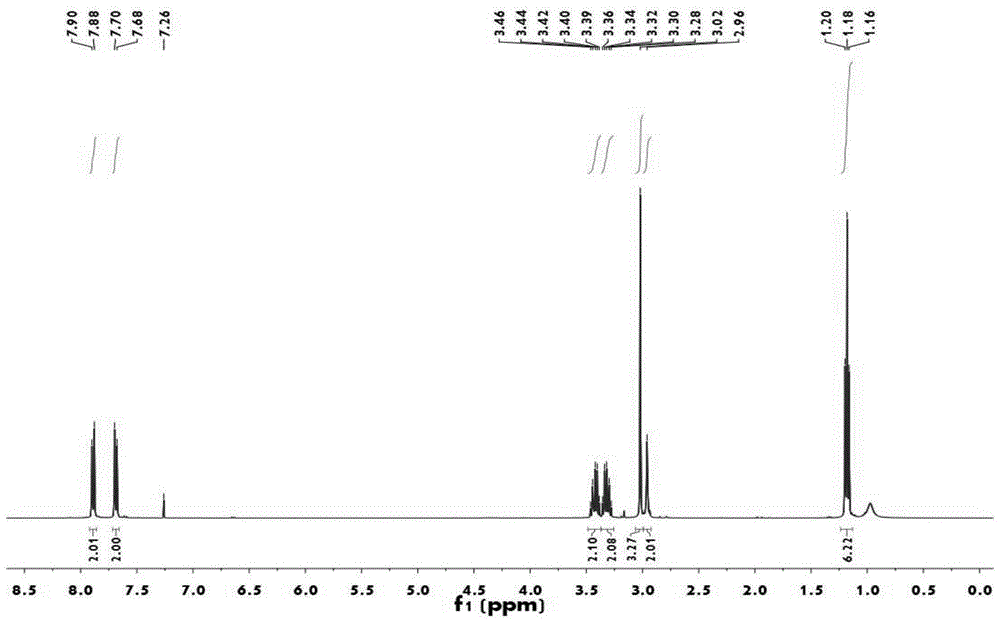
图1为本发明制备的艾瑞昔布中间体(化合物4)的核磁图谱。
图2为本发明制备的艾瑞昔布中间体(化合物5)的核磁图谱。
图3为本发明制备的艾瑞昔布中间体(化合物6)的核磁图谱。
图4为本发明制备的艾瑞昔布(化合物8)的核磁图谱。
具体实施方式
如果无特殊声明,本发明的实施例中采用的原料均为本领域的常用原料,实施例中所采用的方法,均为本领域的常规方法。
缩写词:
pe:石油醚(60-900c);ea:乙酸乙酯;dcm:二氯甲烷;kome(甲醇钾);mtbe:甲基叔丁基醚;et3n:三乙胺;thf:四氢呋喃;edci:碳化二亚胺;hobt:1-羟基苯并三唑;dmap:4-二甲氨基吡啶。
实施例1、本发明艾瑞昔布中间体(化合物4)的制备
本发明艾瑞昔布中间体(化合物4)的合成路线如下:
步骤1:本发明艾瑞昔布中间体(化合物2)的制备
量取20%naoh溶液(60ml)加入250ml圆底烧瓶中,冷却至0℃;再称取nh2oh·hcl(8.4g,1.2mol)溶于10ml水中加入到圆底烧瓶中,最后加入4-甲基亚砜苯乙酮(20g,1mol),冰浴下反应2h。tlc(pe:ea=1:1)监测反应进度,反应完毕后得反应液。
ea萃取反应液4次,每次ea的用量为100ml,合并有机相,无水naso4干燥有机相,再旋干有机相,得20g白色固体(化合物2)。产率93%。
步骤2:本发明艾瑞昔布中间体(化合物3)的制备
向500ml圆底烧瓶中依次加入化合物2(20g,94mmol),对甲基苯磺酰氯(27g,140mmol),85%含量的koh(25g,375mmol),最后加入400mldcm,室温下反应4~5h。tlc(pe:ea=1:1)监测反应进度。反应完毕后,直接抽滤洗涤,旋干滤液得淡黄色固体31g(化合物3),产率90%。
步骤3:本发明艾瑞昔布中间体(化合物4)的制备
称取化合物3(20g,54mmol)加入1l圆底烧瓶中,再加入300ml无水乙醇,充分搅拌,最后称取kome(4.6g,65mmol)溶于50ml无水乙醇中,70℃下反应4~5h。tlc(dcm:meoh=10:1)监测反应进度。反应结束后,向反应液中加入300mlmtbe,有固体析出,抽滤,除去固体,旋去部分滤液,向滤液中通hcl气体半小时,至ph为3;最后旋干滤液。加入100ml乙酸乙酯,再用naoh调节ph至10,ea萃取,旋干有机相。再通过酸碱倒置一次,可得淡黄色固体12g(化合物4),产率75%。
本发明制备所得化合物4的核磁图谱如图1所示:1hnmr(400mhz,cdcl3):δ7.89(d,j=8.5hz,2h),7.69(d,j=8.6hz,2h),3.43(m,2h),3.32(m,2h),3.02(s,3h),2.96(s,2h),1.18(t,j=7.1hz,6h).
本发明制备得到的化合物4基本结构为:
当步骤3中的溶剂为无水乙醇时,得到的是本发明化合物4。当将无水乙醇替换为其他醇溶剂时,可以得到取代基不同的化合物,得到更多的艾瑞昔布中间体。其中,r1、r2分别独立选自氢、取代或未取代的c1~c6烷基;所述取代基为卤素、羟基、氰基、硝基、氨基;优选r1和r2同时为氢、取代或未取代的c1~c6烷基;所述取代基为卤素、羟基、氰基、硝基、氨基。
实施例2、本发明艾瑞昔布中间体(化合物5)的制备
称取化合物4(10g,35mmol)加入250ml圆底烧瓶中,向烧瓶中加入100ml乙腈,接着加入et3n(7.3ml,52mmol),最后在室温下滴加丙酰氯(3.7ml,42mmol),继续在室温下反应1~2h。tlc(dcm:meoh=20:1)监测反应进度。反应结束后,向反应液中加水淬灭反应,旋去乙腈和过量的三乙胺,再用乙酸乙酯萃取,饱和食盐水洗涤有机相,无水硫酸钠干燥。旋干有机相得棕黄色油状液体12g(化合物5),产率100%。
本发明制备所得化合物5的核磁图谱如图2所示:1hnmr(400mhz,cdcl3)δ7.89(d,j=8.1hz,2h),7.70(d,j=8.3hz,2h),5.22(s,1h),3.67(d,j=5.8hz,2h),3.48(m,2h),3.34(m,2h),3.03(s,3h),1.99(q,j=7.5hz,2h),1.19(t,j=7.0hz,6h),0.94(t,j=7.9hz,3h).
实施例3、本发明艾瑞昔布中间体(化合物6)的制备
称取化合物5(10g,29mmol)加入250ml圆底烧瓶中,在n2保护条件下,向烧瓶中加入20mlthf溶剂,80℃下回流,最后再向反应体系中加入1mol/l的硼烷四氢呋喃溶液(58ml,58mmol),继续回流反应1h。tlc(dcm:meoh=10:1)监测反应进度。反应结束后,向反应液中加meoh淬灭反应,旋干反应液,再加入30ml稀盐酸(1mol/l),用乙酸乙酯洗涤3次,每次30ml,水相用20%的naoh溶液调节ph至10,用ea萃取3次,每次50ml,饱和食盐水洗涤有机相,无水硫酸钠干燥。旋干有机相得淡黄色油状液体8g(化合物6),产率83%。
本发明制备所得化合物6的核磁图谱如图3所示:1hnmr(400mhz,cdcl3)δ7.91(d,j=8.5hz,2h),7.74(d,j=8.6hz,2h),3.46(m,2h),3.39-3.28(m,2h),3.05(s,4h),2.98(s,2h),2.41-2.34(m,2h),1.36–1.26(m,2h),1.21(t,j=7.1hz,6h),0.74(t,j=7.4hz,3h).
实施例4、本发明艾瑞昔布的制备
本发明艾瑞昔布(化合物8)的合成路线如下:
步骤6:本发明艾瑞昔布中间体(化合物7)的制备
向250ml圆底烧瓶中,依次加入对甲基苯乙酸(4.5g,30mmol),edci(5.8g,30mmol),hobt(4.0g,30mmol),dmap(183mg,1.5mmol),dcm(100ml),et3n(12.4ml,90mmol),在室温下搅拌反应半小时,最后加入化合物6(5.0g,15mmol),继续在室温下反应1~2h。tlc(pe:ea=1:1)监测反应进度。反应结束后,依次用水(100ml),1nhcl(100ml),饱和nahco3(100ml),饱和食盐水(100ml)洗涤反应液,无水硫酸钠干燥有机相,旋干有机相,再加入浓盐酸(15ml)将产物水解(有大量白色固体析出),抽滤,用水(150ml)洗涤,乙醇重结晶。得类白色固体4.5g(化合物7),产率78%。
步骤7:本发明艾瑞昔布(化合物8)的制备
在50ml圆底烧瓶中依次加入化合物7(3g,7.8mmol),乙醇90ml,水100ml,碳酸钾(2.1g,15.6mmol),搅拌加热至80~120℃回流溶解。反应2h。tlc(pe:ea=1:1)监测反应进度。反应结束后,稍冷至60℃,将反应液倒入150ml冰水中,用1n盐酸中和至中性(ph=6.5-7.0),搅拌冷却至10℃,静置3h,过滤得类白色固体产物2.3g(化合物8,即艾瑞昔布),产率79%。
本发明制备所得化合物8的核磁图谱如图4所示:1hnmr(400mhz,cdcl3)δ7.84(d,j=8.5hz,2h),7.47(d,j=8.5hz,2h),7.27(d,j=6.4hz,2h),7.15(d,j=8.0hz,2h),4.29(s,2h),3.60–3.51(t,j=7.2hz2h),3.05(s,3h),2.35(s,3h),1.70(m,2h),0.98(t,j=7.4hz,3h).[m+h]+calcd.forc21h23no3s,370.1;found,370.1.
对比例1
将本发明实施例1步骤3中通入hcl气体换为通入hcl水溶液,无法得到稳定的化合物,即无法得到本发明艾瑞昔布中间体,也无法通过本发明制备艾瑞昔布的方法制备艾瑞昔布。
综上,本发明提供了一种艾瑞昔布中间体及其制备方法。本发明艾瑞昔布中间体十分稳定;且制备该艾瑞昔布中间体和利用该艾瑞昔布中间体制备艾瑞昔布的过程中,反应条件温和,试剂毒性小,对环境友好;此外,反应过程中产物易分离纯化,且产率高,适合大规模的工业生产。
- 还没有人留言评论。精彩留言会获得点赞!