一种改变菊花香气成分萜烯类挥发物含量的方法与流程
本发明属于植物基因工程和转基因育种领域,涉及一种通过转cmwrky7基因改变菊花香气成分萜烯类挥发物含量的方法。
背景技术:
菊花(chrysanthemummorifolium)起源于我国,是我国十大传统名花和世界四大切花之一,产量居四大切花之首,在生产和园林应用中占有十分重要的地位,可做观赏、药用、食用等,具有很高的观赏和经济价值。
菊花挥发油主要为β-蒎烯、桉叶油醇、樟脑、龙脑、乙酸龙脑酯等化合物,上述化合物均为单萜或单萜衍生物(zhangetal.,2010)。因此,萜类及其衍生物特别是单萜或单萜衍生物为菊花花香的主要成分,对其合成路径的调控能够影响菊花的花香。
wrky是植物所特有的一类转录因子,是近年来新发现的新型锌指型转录调控因子。wrky是一个庞大的转录因子家族,家族成员在结构上有共同的特点:每个成员都含有一个wrky结构域,该结构域是由一段大约60个高度保守的氨基酸残基所组成的多肽序列。另外,wrky转录因子的dna结合域中一般都含有一个锌指结构(rushtonetal.,1996)。迄今,在拟南芥(arabidopsisthaliana)中共克隆了74个wrky成员(eulgem,2005),而水稻中则发现了105个完整的wrky家族成员(muthamilarasanetal.,2015)。wrky转录因子参与植物多个信号传导途径的调控,如植物的生长发育、对生物胁迫和非生物胁迫的应激响应等,其表达具有快速、瞬时等特点(wangetal.,2016,dingetal.,2016,tripathietal.,2014,wuetal.,2016),同时还具有不同发育阶段与组织特异性表达特性(tripathietal.,2014)。通常情况下,wrky转录因子的下游基因启动子序列中会出现多个w-box,wrky蛋白可以特异地与之结合从而调节相关基因的表达,(ciolkowskietal.,2008)。
研究表明,wrky可以调控植物萜类物质的合成,不仅可以直接调控萜类合成酶基因的表达,而且可以参与萜类物质合成路径中的一系列基因的表达调控(maetal.,2009)。如棉花gawrky1可以调控杜松烯合成酶基因(gacad1-a)的表达(xuetal.,2004);长春花crwrky1可以调控萜类生物碱的合成(suttipantaetal.,2011);青蒿aawrky1不仅可以调控紫槐烯合酶基因(aaads)的表达,还可以调节萜类合成路径中一系列基因的表达(maetal.,2009)。由于萜类代谢途径中一系列合成酶在转录水平上往往呈现出相关性,这些合成酶的表达可能受到同一个/类转录因子的调控。因此,wrky转录因子家族基因很有可能也参与菊花萜类合成路径的调控。
参考文献:
ciolkowskii,wanked,birkenbihlr,somssichi,2008.studiesondna-bindingselectivityofwrkytranscriptionfactorslendstructuralcluesintowrky-domainfunction.plantmolecularbiology68,81-92.
dingd,fangw,shis,zhaoy,lix,xiaok,2016.wheatwrkytypetranscriptionfactorgenetawrky1isessentialinmediatingdroughttoleranceassociatedwithanaba-dependentpathway.plantmolecularbiologyreporter34,1111–1126.
eulgemt,2005.regulationofthearabidopsisdefensetranscriptome.trendsinplantscience10,71-8.
mad,pug,leic,etal.,2009.isolationandcharacterizationofaawrky1,anartemisiaannuatranscriptionfactorthatregulatestheamorpha-4,11-dienesynthasegene,akeygeneofartemisininbiosynthesis.plantandcellphysiology50,2146-61.
muthamilarasanm,bonthalav,khandelwalr,etal.,2015.globalanalysisofwrkytranscriptionfactorsuperfamilyinsetariaidentifiespotentialcandidatesinvolvedinabioticstresssignaling.frontiersinplantscience6,910.
rushtonp,torresj,parniskem,wernertp,hahlbrockk,somssichi,1996.interactionofelicitor-induceddna-bindingproteinswithelicitorresponseelementsinthepromotersofparsleypr1genes.theembojournal15,5690–5700.
suttipantan,pattanaiks,kulshrestham,patrab,singhsk,yuanl,2011.thetranscriptionfactorcrwrky1positivelyregulatestheterpenoidindolealkaloidbiosynthesisincatharanthusroseus.plantphysiology157,2081-93.
tripathip,rabarar,rushtonp,2014.asystemsbiologyperspectiveontheroleofwrkytranscriptionfactorsindroughtresponsesinplants.planta239,255-66.
wangs,wangj,yaow,zhoub,jiangt,2016.expressionpatternsofwrkygenesindi-haploidpopulussimonii×p.nigrainresponsetoosmoticstressandabatreatment.plantgrowthregulation78,325-333.
wuz,lix,liuz,lih,wangy,zhuangj,2016.transcriptome-wideidentificationofcamelliasinensiswrkytranscriptionfactorsinresponsetotemperaturestress.moleculargeneticsandgenomics291,255-269.
xuy,wangj,wangs,wangj,chenx,2004.characterizationofgawrky1,acottontranscriptionfactorthatregulatesthesesquiterpenesynthasegene(+)-δ-cadinenesynthase-a.plantphysiology135,507-15.
zhangc,qinm,shup,hongj,lül,hed,2010.chemicalvariationsoftheessentialoilsinflowerheadsofchrysanthemumindicuml.fromchina.chemistryandbiodiversity7,2951-62.
技术实现要素:
本发明的目的在于解决定向改变菊花香气成分萜烯类挥发物含量的问题,提供cmwrky7基因在调控菊花香气成分萜烯类挥发物含量中的基因工程应用。
本发明的另一目的是提供一种通过转cmwrky7基因改变菊花香气成分萜烯类挥发物含量的方法。本发明涉及的农杆菌介导遗传转化,转基因植物的分子检测及转基因植株后代gc-ms检测观察表型用于本发明,为利用基因工程技术开展芳香菊花分子育种提供方法和实例。
本发明的又一目的在于提供一种cmwrky7基因植物表达载体,及该载体在降低菊花香气成分萜烯类挥发物含量中的基因工程应用。
本发明的再一目的是提供cmwrky7基因或cmwrky7基因植物表达载体改变菊花香气中的基因工程应用。
本发明的目的可以通过以下技术方案实现:
cmwrky7基因在调控菊花香气成分萜烯类挥发物含量中的基因工程应用,所述cmwrky7基因的核苷酸序列如seqidno.1所示:
atgtgcactgatgatgggttagagggtagtgttgatgatggttacagctggagaaagtatggacagaaagacattttgggtgccaaatttccaaggagctattatagatgcacataccgcaaggctgagaagtgcttggcgacaaaacaagtacagagaaccgatgcaaatcccacagtatttgatatcacatacaaaggaaaacacacttgcaaccatcacgctcgtttggccgaaccacctttgcctgaaaaacatgaaataaacacaacccaccaccaacaactatcacgaccgaatccgggtgaaatgctttcaaatctgagagccaacctcgctgttaacacctcggattttggtgctactgacccatactcattttccttccctttagaaccatttggcatcattgaagattaccaacaacttcatttgcctaatgattttgatgatgaattgttgcaagtctattcgccacctttgatctccccgggcacttctgattcaaactgctacacggactgggacagttcaccatcactaaacttcacggtatatttagaccatgattttaaattt
进一步的,所述的应用为,将所述cmwrky7基因植物表达载体导入切花菊,降低菊花香气成分萜烯类挥发物含量。
进一步的,所述的应用为,所述萜烯类为单萜和/或单萜衍生物。
进一步的,所述cmwrky7基因植物表达载体的构建方法为:以茶用菊‘滁菊’的cdna为模板,设计如seqidno.4所示的上游引物cmwrky7-bamhi-f和seqidno.5所示的下游引物cmwrky7-noti-r,进行pcr反应,在目的基因cmwrky7的上游和下游分别引入bamhi和noti酶切位点;将pcr产物连接到pmd19-t载体,转化dh5α感受态细胞,提取阳性质粒,将提取的阳性质粒由bamhi和noti双酶切得到的cmwrky7片段与bamhi和noti双酶切的pentrtm1a连接,转化,提取的阳性质粒经pvui单酶切线性化后与植物表达载体pmdc43质粒进行lr重组反应,转化,得到cmwrky7基因植物表达载体pmdc43-cmwrky7,所述的cmwrky7基因的序列为seqidno.1。
上游引物cmwrky7-bamhi-f:5'-cgcggatccggatgtgcactgatgatgggtt-3'(seqidno.4)
下游引物cmwrky7-noti-r:5'-atttgcggccgcaaaatttaaaatcatggtcta-3'(seqidno.5)
一种通过转cmwrky7基因改变菊花香气成分萜烯类挥发物含量的方法,所述方法包括以下步骤:
(1)以茶用菊‘滁菊’为材料,提取总rna,反转录获得cdna;
(2)构建cmwrky7基因植物表达载体:以步骤(1)获得的cdna为模板,设计如seqidno.4所示的上游引物cmwrky7-bamhi-f和seqidno.5所示的下游引物cmwrky7-noti-r,进行pcr反应,在目的基因cmwrky7的上游和下游分别引入bamhi和noti酶切位点;将pcr产物连接到pmd19-t载体,转化dh5α感受态细胞,提取阳性质粒,将提取的阳性质粒由bamhi和noti双酶切得到的cmwrky7片段与bamhi和noti双酶切的pentrtm1a连接,转化,提取的阳性质粒经pvui单酶切线性化后与植物表达载体pmdc43质粒进行lr重组反应,转化,得到cmwrky7基因植物表达载体pmdc43-cmwrky7;
(3)将步骤(2)构建得到的cmwrky7基因植物表达载体加入感受态农杆菌eha105,挑选阳性克隆,通过叶盘法转化菊花,将pmdc43-cmwrky7导入菊花,对初步获得潮霉素抗性的转基因植株进行pcr鉴定、荧光定量rt-pcr分子检测,验证内源基因已经转入转基因植株的基因组dna中并表达,筛选阳性转cmwrky7基因植株,即为改变菊花香气成分萜烯类挥发物含量的转基因菊花植株。
进一步的,步骤(3)中所述对初步获得潮霉素抗性的转基因植株进行pcr鉴定、荧光定量rt-pcr分子检测的步骤为:
所述pcr鉴定为:
取筛选得到的潮霉素抗性植株嫩叶及未转化株嫩叶,提取基因组dna,设计扩增反应引物,扩增出的片段长为750bp,引物序列为:
上游引物pmdc43-hyg-f:5′-cttctacacagccatcggtccag-3′(seqidno.6),
下游引物pmdc43-hyg-r:5′-cggaagtgcttgacattggggag-3′(seqidno.7);
分别以潮霉素抗性植株和未转化植株dna为模板,以pmdc43-gfp-f和pmdc43-gfp-r为引物,进行pcr检测,扩增产物进行琼脂糖凝胶电泳检测分析;
所述的荧光定量rt-pcr分子检测为:
提取潮霉素抗性植株及未转化株总rna,反转录成第一链cdna,建立荧光定量rt-pcr扩增体系,根据数据分析得到各个样品的ct值,以未转化植株的表达量为基准值,计算各转基因植株及野生型基因相对表达情况;特异引物扩增出的片段长为105bp,引物序列为:
上游引物cmwrky7-rt-f:5′-cgggtgaaatgctttcaaat-3′(seqidno.8),
下游引物cmwrky7-rt-r:5′-tgccaaatggttctaaaggg-3′(seqidno.9);
以ef1α扩增的基因片段为内标,片段长度为151bp,引物序列:
上游引物ef1α-f:5′-ttttggtatctggtcctggag-3′(seqidno.10),
下游引物ef1α-r:5′-ccattcaagcgacagactca-3′(seqidno.11);
一种cmwrky7基因植物表达载体,所述cmwrky7基因植物表达载体通过以下方法制备:以茶用菊‘滁菊’的cdna为模板,设计如seqidno.4所示的上游引物cmwrky7-bamhi-f和seqidno.5所示的下游引物cmwrky7-noti-r,进行pcr反应,在目的基因cmwrky7的上游和下游分别引入bamhi和noti酶切位点;将pcr产物连接到pmd19-t载体,转化dh5α感受态细胞,提取阳性质粒,将提取的阳性质粒由bamhi和noti双酶切得到的cmwrky7片段与bamhi和noti双酶切的pentrtm1a连接,转化,提取的阳性质粒经pvui单酶切线性化后与植物表达载体pmdc43质粒进行lr重组反应,转化,得到cmwrky7基因植物表达载体pmdc43-cmwrky7。
上述的cmwrky7基因植物表达载体在降低菊花香气成分萜烯类挥发物含量中的基因工程应用。
进一步的,采用农杆菌介导的叶盘法将所述的cmwrky7基因植物表达载体导入菊花,降低菊花香气成分萜烯类挥发物含量。
进一步的,所述萜烯类为单萜和/或单萜衍生物。
如seqidno.1所示的cmwrky7基因或前述的cmwrky7基因植物表达载体改变菊花香气中的基因工程应用,
进一步的,所述的应用为,将如seqidno.1所示的cmwrky7基因或前述的cmwrky7基因植物表达载体导入菊花,降低菊花花香气成分萜烯类挥发物含量,从而改变菊花香气。
本发明所述的技术方案利用根癌农杆菌介导法,将cmwrky7基因导入切花菊,并经过潮霉素筛选抗性植株;经过潮霉素抗性筛选得到阳性转化植株,对转化植株进行pcr、定量rt-pcr检测验证内源基因已经转入转基因植株的基因组dna中并发生转录。对转基因植株后代进行表型观察,最终获得菊花香气成分萜烯类挥发物含量改变的转基因菊花植株。cmwrky7基因从‘滁菊’中克隆得到,该基因的序列为seqidno.1。
本发明所述的通过转cmwrky7基因改变菊花萜烯类挥发物含量的方法,该方法包括以下步骤:
(1)菊花cmwrky7基因的克隆
以茶用菊‘滁菊’为材料,提取rna并反转录成cdna,根据序列信息设计特异引物,结合pcr扩增目的基因的全长序列,
上游引物cmwrky7-orf-f:5'-atgtgcactgatgatgggttag-3'(seqidno.2)
下游引物cmwrky7-orf-r:5'-ttaaaatttaaaatcatggtctaaata-3'(seqidno.3)
以反转录的cdna为模板,进行pcr反应,产物连接到pmd19-t载体,转化dh5α感受态细胞,获得目的基因的序列为seqidno.1。
(2)植物表达载体pmdc43-cmwrky7的构建
根据cmwrky7基因全长序列设计引物,以茶用菊‘滁菊’cdna为模板,用高保真酶进行pcr反应,在cmwrky7基因的上游和下游分别引入bamhi和noti酶切位点,
上游引物cmwrky7-bamhi-f:5'-cgcggatccggatgtgcactgatgatgggtt-3'(seqidno.4)
下游引物cmwrky7-noti-r:5'-atttgcggccgcaaaatttaaaatcatggtcta-3'(seqidno.5)
pcr产物连接到pmd19-t载体,转化dh5α感受态细胞,提取阳性质粒,将提取的阳性质粒由bamhi和noti双酶切得到的cmwrky7片段与bamhi和noti双酶切的pentrtm1a连接,转化,提取的阳性质粒经pvui单酶切线性化后与植物表达载体pmdc43质粒进行lr重组反应,转化,提取阳性质粒,植物表达载体pmdc43-cmwrky7构建成功,所述的cmwrky7基因序列为seqidno.1。
(3)采用农杆菌介导法将步骤(2)构建的cmwrky7基因植物表达载体转入菊花‘神马’中,进行培养初步获得抗性植株。
制备感受态的农杆菌,将步骤(2)中构建的cmwrky7基因植物表达载体转入感受态的农杆菌中,挑选阳性克隆,摇菌至od=0.5,离心后,弃上清,用ms(ph5.8)培养液将沉淀等体积悬浮,用于侵染;取组培瓶中菊花苗顶端叶盘作为转化受体,在预培养基中培养3d,然后浸入上述备好的转入了pmdc43-cmwrky7的植物表达载体的农杆菌菌液中侵染8~10min,用滤纸吸干叶盘表面的菌液后再将其接种到共培养基上,暗培养3d,然后转入筛选培养基上继代培养4代,等分化出的抗性芽长至2~3厘米时转入生根培养基上培养,初步获得抗性植株。所用的农杆菌菌株为eha105。
菊花组织培养基以ms培养基为基础,ph5.8,100kpa、121℃灭菌20分钟;预培养培养基:ms+6-苄氨基嘌呤(6-ba)1mg/l+萘乙酸(naa)0.5mg/l;共培养培养基:ms+6-苄氨基嘌呤(6-ba)1mg/l+萘乙酸(naa)0.5mg/l;筛选培养基:ms+潮霉素(hyg)10mg/l+羧卞青霉素(carb)500mg/l+6-苄氨基嘌呤(6-ba)1mg/l+萘乙酸(naa)0.1mg/l;生根培养基:ms+潮霉素(hyg)8mg/l。
详细操作过程为:
从yeb(50μg/ml利福平)平板上挑取eha105单菌落,接种于50ml含50μg/ml利福平的yeb液体培养基中,200rpm,28℃培养至od值0.5,而后冰浴菌液30min,离心收集菌体,悬浮于2ml预冷的100mmcacl2(20%甘油)溶液中,200μl/管分装,待用。
取10μlpmdc43-cmwrky7载体质粒,加入200μl感受态细胞,冰浴30min,液氮冷冻5min,37℃5min,加入800μlyeb液体培养基,28℃200rpm预培养4h,菌液涂板于yeb(50μg/ml利福平+50μg/ml卡那霉素)固体培养基上,28℃暗培养2天,挑取单克隆检测,选取阳性克隆摇菌,用于转化菊花。
以切花菊‘神马’叶片为外植体,取组培瓶中菊花苗顶端叶盘(0.5cmⅹ0.5cm)作为转化受体,在预培养培养基中预培养3d,然后浸入备好的农杆菌菌液中感染8~10min,用滤纸吸干菌液后再接种到共培养基上黑暗中共培养3d,然后转入筛选培养基上继代培养4代,等分化出的抗性芽长至2~3厘米时转入生根培养基上培养,初步获得抗性植株。
将转cmwrky7基因初步获得的抗性植株进行pcr鉴定、荧光定量rt-pcr分子检测,筛选阳性植株,获得转基因菊花株系:
(1)pcr检测
取生根筛选得到的潮霉素抗性植株嫩叶及未转化株嫩叶,提取基因组dna,将潮霉素抗性基因作为检测目标,根据潮霉素抗性基因两端合成扩增反应引物,扩增出的片段长为750bp,引物序列为:
上游引物pmdc43-hyg-f:5′-cttctacacagccatcggtccag-3′(seqidno.6),
下游引物pmdc43-hyg-r:5′-cggaagtgcttgacattggggag-3′(seqidno.7);
分别以潮霉素抗性植株和未转化植株dna为模板,以pmdc43-gfp-f和pmdc43-gfp-r为引物,进行pcr检测,扩增产物进行琼脂糖凝胶电泳检测分析;
(2)荧光定量rt-pcr检测
提取抗潮霉素植株及未转化株总rna,反转录成第一链cdna,建立荧光定量rt-pcr扩增体系,每个样品重复3次,根据数据分析得到各个样品的ct值,以未转化植株的表达量为基准值,计算各转基因植株及野生型基因相对表达情况;特异引物扩增出的片段长为105bp,引物序列为:
上游引物cmwrky7-rt-f:5′-cgggtgaaatgctttcaaat-3′(seqidno.8),
下游引物cmwrky7-rt-r:5′-tgccaaatggttctaaaggg-3′(seqidno.9);
以ef1α扩增的基因片段为内标,片段长度为151bp,引物序列:
上游引物ef1α-f:5′-ttttggtatctggtcctggag-3′(seqidno.10),
下游引物ef1α-r:5′-ccattcaagcgacagactca-3′(seqidno.11);
对通过pcr、荧光定量rt-pcr分子检测所获得转基因植株后代进行表型观察:
选取6-8叶龄的野生型菊花(wt)和转基因菊花(ox20)的扦插苗作为表型观察的材料,每个株系10株苗。乙酸乙酯提取每个株系的挥发物成分,并利用gc-ms检测每个株系的萜烯类物质的含量。
本发明所提供的改变菊花香气成分萜烯类挥发物含量的方法具有如下优点:
本发明提供的方法选育了转基因菊花材料,采用转基因技术,将cmwrky7基因整合到菊花基因组中,通过gc-ms分析获得萜烯类物质含量发生改变的转基因菊花株系。结果发现,与未转基因植株相比,转入cmwrky7基因的植株萜烯类物质含量发生变化,进而改变转基因菊花的香气。
附图说明
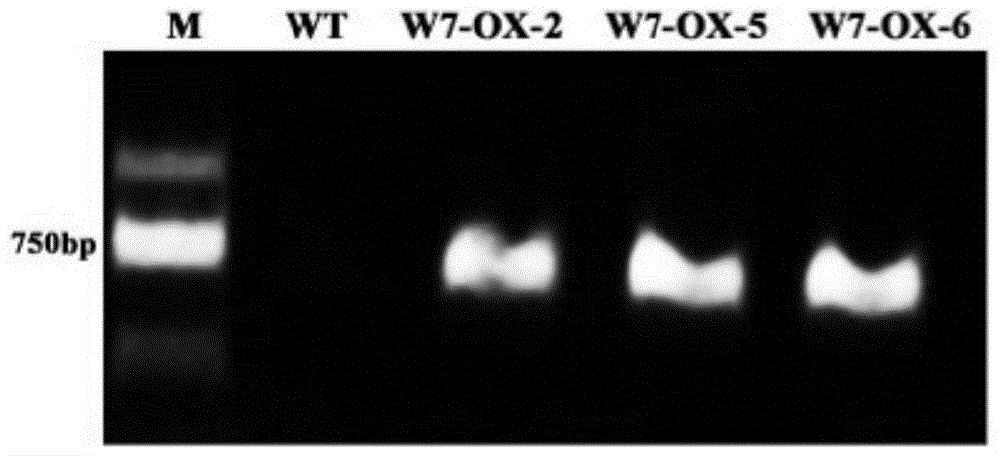
图1转cmwrky7基因菊花特异引物检测电泳
m:dl2000marker;
wt:野生型植株;
w7-ox-2,5,6:转cmwrky7的菊花株系
图2转基因及野生型菊花植株中cmwrky7基因的相对表达量水平
wt:野生型植株;
w7-ox-2,5,6:转cmwrky7的菊花株系
图3野生型及转基因菊花的组培苗
wt:野生型植株;
w7-ox-2,5,6:转cmwrky7的菊花株系
图4转基因及野生型菊花植株中单萜及单萜衍生物类物质的绝对含量
wt:野生型植株;
w7-ox-2,5,6:转cmwrky7的菊花株系
具体实施方式
下面结合实施例对本发明做进一步说明,下列实施例中未注明具体条件的实验方法,通常按照本领域的公知手段。本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,其具体实施方式如下:
实施例1.cmwrky7基因的克隆
以茶用菊‘滁菊’为材料,取0.15g,参照trizolrna提取试剂盒(takara)说明书的操作方法提取总rna,根据m-mlv反转录试剂盒(takara)进行反转录获得cdna,根据菊花文库中该基因的序列信息,运用primer5软件设计特异引物扩增cmwrky7;
上游引物cmwrky7-orf-f:5'-atgtgcactgatgatgggttag-3'(seqidno.2)
下游引物cmwrky7-orf-r:5'-ttaaaatttaaaatcatggtctaaata-3'(seqidno.3)
以获得的cdna为模板,进行pcr反应,50μl反应体系:10×pcrbuffer5.0μl,cmwrky7-orf-f、cmwrky7-orf-r引物各1.0μl(20μmol·l-1),dntpmix4.0μl(2.5mmol·l-1),taqdnapolymerase0.2μl,cdna模板1μl,ddh2o37.8μl;反应程序:95℃预变性5min,然后94℃解链45sec,55℃退火45sec,72℃延伸1min,反应30个循环,72℃延伸10min;产物用凝胶回收试剂盒(axygen,usa)回收,用t4dna连接酶(takara)连接到pmd19-t载体(takara),转化dh5α感受态细胞,序列测定为seqidno.1:
atgtgcactgatgatgggttagagggtagtgttgatgatggttacagctggagaaagtatggacagaaagacattttgggtgccaaatttccaaggagctattatagatgcacataccgcaaggctgagaagtgcttggcgacaaaacaagtacagagaaccgatgcaaatcccacagtatttgatatcacatacaaaggaaaacacacttgcaaccatcacgctcgtttggccgaaccacctttgcctgaaaaacatgaaataaacacaacccaccaccaacaactatcacgaccgaatccgggtgaaatgctttcaaatctgagagccaacctcgctgttaacacctcggattttggtgctactgacccatactcattttccttccctttagaaccatttggcatcattgaagattaccaacaacttcatttgcctaatgattttgatgatgaattgttgcaagtctattcgccacctttgatctccccgggcacttctgattcaaactgctacacggactgggacagttcaccatcactaaacttcacggtatatttagaccatgattttaaattt
实施例2.植物表达载体pmdc43-cmwrky7的构建
以茶用菊‘滁菊’的cdna为模板,根据cmwrky7全长基因序列设计如seqidno.4所示的上游引物cmwrky7-bamhi-f和seqidno.5所示的下游引物cmwrky7-noti-r,引物进行pcr反应,在目的基因cmwrky7的上游和下游分别引入酶切位点bamhi和noti,pcr产物连接到pmd19-t载体,转化dh5α感受态细胞,提取阳性质粒,将提取的阳性质粒用bamhi和noti双酶切得到的cmwrky7片段与bamhi和noti双酶切的pentrtm1a连接,转化,提取的阳性质粒经pvui单酶切线性化后与植物表达载体pmdc43质粒进行lr重组反应(invitrogen,usa),转化,提取阳性质粒。
上游引物cmwrky7-bamhi-f:5'-cgcggatccggatgtgcactgatgatgggtt-3'(seqidno.4)
下游引物cmwrky7-noti-r:5'-atttgcggccgcaaaatttaaaatcatggtcta-3'(seqidno.5)
以‘滁菊’cdna作为模板,用高保真酶(primestartmhsdnapolymerase,takara)进行pcr反应,50μl反应体系:10×hspcrbuffer5.0μlcmwrky7-bamhi-f、cmwrky7-noti-r引物各1.0μl(20μmol·l-1),dntpmix4.0μl(2.5mmol·l-1),primestartmhsdnapolymerase0.4μl,cdna模板1μl,ddh2o37.6μl;反应程序:95℃预变性5min,然后94℃解链45sec,55℃退火30sec,72℃延伸1min,反应30个循环,72℃延伸10min;pcr产物用凝胶回收试剂盒(axygen,usa)回收,连接到pmd19-t载体,转化dh5α感受态细胞,提取阳性质粒pmd19-cmwrky7;
取gateway入门载体pentrtm1a(invitrogen,usa)和pmd19-cmwrky7用bamhi和noti双酶切,双酶切体系(50μl):10×hbuffer5μl,bsa5μl,质粒pentrtm1a或pmd19-cmwrky715μl,bamhi2μl,noti2μl,ddh2o21μl;37℃反应1h;双酶切产物进行琼脂糖凝胶电泳分析,用凝胶回收试剂盒(axygen)回收质粒pentrtm1a大片段和cmwrky7片段。用t4dna连接酶(takara)连接两个回收的产物,连接反应体系(10μl):10×t4ligasebuffer1μl,pentrtm1a大片段2μl,cmwrky7小片段6μl,t4dna连接酶1μl;16℃过夜连接反应,取5μl连接产物转化dh5α感受态细胞。37℃过夜培养,挑取阳性单克隆扩大培养,提取质粒pentrtm1a-cmwrky7。
取质粒pentrtm1a-cmwrky7用pvui单酶切,酶切体系(20μl):10×rebuffer2μl,acetylatedbsa2μl,质粒pentrtm1a-cmwrky710μl,pvui1μl,ddh2o5μl;37℃反应2h,用凝胶回收试剂盒(axygen)回收质粒pentrtm1a-cmwrky7大片段。将回收的片段与pmdc43载体质粒进行lr重组反应,反应体系(5μl):pentrtm1a-cmwrky7大片段3μl,pmdc43质粒1μl,lrclonasetmⅱenzymemix1μl;25℃反应1h,加入1μlproteinasek,37℃反应10min;取5μl重组产物转化dh5α感受态细胞,37℃过夜培养,挑取阳性单克隆扩大培养,提取质粒pmdc43-cmwrky7,电泳和测序验证。植物表达载体pmdc43-cmwrky7的构建成功。
实施例3.农杆菌eha105介导叶盘法转化菊花
从yeb(50μg/ml利福平)平板上挑取eha105单菌落,接种于50ml含50μg/ml利福平的yeb液体培养基中,200rpm,28℃培养至od值0.5,而后冰浴菌液30min,离心收集菌体,悬浮于2ml预冷的100mmcacl2(20%甘油)溶液中,200μl/管分装,待用。取10μlpmdc43-cmwrky7载体质粒,加入200μl感受态细胞,冰浴30min,液氮冷冻5min,37℃5min,加入800μlyeb液体培养基,28℃200rpm预培养4h,菌液涂板于yeb(50μg/ml利福平+50μg/ml卡那霉素)固体培养基上,28℃暗培养2天,挑取单克隆检测,选取阳性克隆摇菌,用于转化菊花。
采用的农杆菌指包含cmwrky7基因的植物表达载体的eha105,用yeb液体培养基培养农杆菌eha105;将转基因植株后代命名为ox20-n,以切花菊‘神马’叶片为外植体,采用根癌农杆菌介导法将‘滁菊’中克隆得到的cmwrky7基因导入菊花。取组培瓶中菊花苗顶端叶盘(0.5cmⅹ0.5cm)作为转化受体,在预培养培养基中预培养3d,然后浸入备好的农杆菌菌液中感染10min,用滤纸吸干菌液后再接种到共培养基上黑暗中共培养3d,然后转入筛选培养基上继代培养4代,等分化出的抗性芽长至2~3厘米时转入生根培养基上培养,初步获得抗性植株(图3)。
菊花组织培养基以ms培养基为基础,ph5.8,100kpa、121℃灭菌20分钟;预培养培养基:ms+6-苄氨基嘌呤(6-ba)1mg/l+萘乙酸(naa)0.5mg/l;共培养培养基:ms+6-苄氨基嘌呤(6-ba)1mg/l+萘乙酸(naa)0.5mg/l;筛选培养基:ms+潮霉素(hyg)10mg/l+羧卞青霉素(carb)500mg/l+6-苄氨基嘌呤(6-ba)1mg/l+萘乙酸(naa)0.1mg/l;生根培养基:ms+潮霉素(hyg)8mg/l。
实施例4.将转cmwrky7基因抗性植株进行分子检测(pcr、荧光定量rt-pcr)呈阳性,获得转基因菊花株系
1)pcr检测
取生根筛选得到的潮霉素抗性植株嫩叶及未转化株嫩叶,提取基因组dna,将潮霉素抗性基因作为检测目标,根据潮霉素抗性基因两端合成扩增反应引物,扩增出的片段长为750bp,引物序列为:
上游引物pmdc43-hyg-f:5′-cttctacacagccatcggtccag-3′(seqidno.6),
下游引物pmdc43-hyg-r:5′-cggaagtgcttgacattggggag-3′(seqidno.7);
分别以潮霉素抗性植株和未转化植株dna为模板,以pmdc43-hyg-f和pmdc43-hyg-r为引物,进行pcr检测。扩增体系为:1μldna模板,2.5μl10×pcrbuffer,2μldntp,1.5μl2.5mmol·l-1mgcl2,5u·μl-1taq酶0.2μl,pmdc43-hyg-f和pmdc43-hyg-r各1μl,去离子水补足体积至25μl。扩增条件为:94℃预变性5min,94℃变性1min,58℃退火45s,72℃延伸1min,30个循环;72℃延伸10min。扩增产物进行琼脂糖凝胶电泳检测分析(如图1)。图中可以看到,转cmwrky7的植株均可扩增出与阳性对照相同的特异扩增带野生型植株没有扩增出条带。
2)荧光定量rt-pcr检测
提取抗潮霉素植株叶片及未转化株叶片总rna,反转录成第一链cdna,用荧光定量rt-pcr对cmwrky7基因的表达量进行检测。按照荧光定量试剂盒(
上游引物cmwrky7-rt-f:5′-cgggtgaaatgctttcaaat-3′(seqidno.8),
下游引物cmwrky7-rt-r:5′-tgccaaatggttctaaaggg-3′(seqidno.9);
以ef1α扩增的基因片段为内标,片段长度为151bp,引物序列:
上游引物ef1α-f:5′-ttttggtatctggtcctggag-3′(seqidno.10),
下游引物ef1α-r:5′-ccattcaagcgacagactca-3′(seqidno.11);
根据荧光定量rt-pcr检测结果(图2),与野生型相比,转cmwrky7的植株株系基因表达量有所提高,w7-ox-2、w7-ox-5、w7-ox-6株系表达量明显较高。证实外源基因已经转入菊花基因组dna中并表达。
实施例5.转基因植株后代的表型观察
取转基因株系w7-ox-2、w7-ox-5、w7-ox-6及野生型菊花组织各0.2g,液氮速冻后利用高速样品研磨机淹没至粉末状,加入1ml含0.002%乙酸壬酯的乙酸乙酯于25℃摇床中萃取2h。高速离心1min,取上清加入2ml样品瓶中待用。样品通过气相联用质谱分析菊花组织中的萜烯成分以及含量,gc-ms色谱条件如下:气相色谱柱尺寸为30m×0.25mm×0.25μm。以流速为1ml·min-1的纯净氦气作为载体,采用不分流式注射(注射温度为250℃)。梯度升温方式,以5℃·min-1的速度从40℃上升到250℃,高温状态下保持20min,试验保留时间为42min。物质的定性分析通过比较nist(nationalinstituteofstandardsandtechnology)质谱数据库中物质的保留时间及标准样的质谱来鉴定。
结果表明:相对于野生型,w7-ox-2、w7-ox-5、w7-ox-6三个超表达转基因株系的多种单萜含量以及多种单萜衍生物含量显著下降(图4)。
可以知道,上述实施例仅为了说明发明原理而采用的示例性实施方式,然而本发明不仅限于此,本领域技术人员在不脱离本发明实质情况下,可以做出各种改进和变更,这些改进和变更也属于本发明的保护范围。
序列表
<110>南京农业大学
<120>一种改变菊花香气成分萜烯类挥发物含量的方法
<160>11
<170>siposequencelisting1.0
<210>1
<211>579
<212>dna
<213>菊花‘滁菊’(chrysanthemummorifolium‘chuju’)
<400>1
atgtgcactgatgatgggttagagggtagtgttgatgatggttacagctggagaaagtat60
ggacagaaagacattttgggtgccaaatttccaaggagctattatagatgcacataccgc120
aaggctgagaagtgcttggcgacaaaacaagtacagagaaccgatgcaaatcccacagta180
tttgatatcacatacaaaggaaaacacacttgcaaccatcacgctcgtttggccgaacca240
cctttgcctgaaaaacatgaaataaacacaacccaccaccaacaactatcacgaccgaat300
ccgggtgaaatgctttcaaatctgagagccaacctcgctgttaacacctcggattttggt360
gctactgacccatactcattttccttccctttagaaccatttggcatcattgaagattac420
caacaacttcatttgcctaatgattttgatgatgaattgttgcaagtctattcgccacct480
ttgatctccccgggcacttctgattcaaactgctacacggactgggacagttcaccatca540
ctaaacttcacggtatatttagaccatgattttaaattt579
<210>2
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>2
atgtgcactgatgatgggttag22
<210>3
<211>27
<212>dna
<213>人工序列(artificialsequence)
<400>3
ttaaaatttaaaatcatggtctaaata27
<210>4
<211>31
<212>dna
<213>人工序列(artificialsequence)
<400>4
cgcggatccggatgtgcactgatgatgggtt31
<210>5
<211>33
<212>dna
<213>人工序列(artificialsequence)
<400>5
atttgcggccgcaaaatttaaaatcatggtcta33
<210>6
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>6
cttctacacagccatcggtccag23
<210>7
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>7
cggaagtgcttgacattggggag23
<210>8
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>8
cgggtgaaatgctttcaaat20
<210>9
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>9
tgccaaatggttctaaaggg20
<210>10
<211>21
<212>dna
<213>人工序列(artificialsequence)
<400>10
ttttggtatctggtcctggag21
<210>11
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>11
ccattcaagcgacagactca20
- 还没有人留言评论。精彩留言会获得点赞!