一种水溶性过渡金属配合物催化分子氧氧化水相合成不对称硫化物的方法与流程
本发明涉及不对称硫化物合成技术领域,具体涉及一种水溶性过渡金属配合物催化分子氧氧化水相合成不对称硫化物的方法。
背景技术:
含硫化合物是重要的有机合成中间体,广泛用于药物化学和材料化学。含硫脲的有机支架化合物,对癌症、疟疾、阿尔茨海默病、帕金森病、艾滋病等疾病疗效显著,在生物和制药领域具有重要影响。含硫化合物也是几种天然产品的基石,例如从印尼海鞘中分离得到多硫芳香族生物碱可以抑制人体结肠腺癌细胞、子宫颈腺癌细胞、乳腺腺癌细胞等癌细胞的生长。含硫化合物在材料科学中也有着广泛的应用,例如对pvc进行内增塑从而取得了良好的塑化效率。有机硫化合物在有机合成中不仅作为生物活性物质和功能材料,而且作为有用的建筑材料或试剂,具有重要的地位。
传统的芳基硫键形成方法是通过硫醇和卤代物加成-消除机理进行取代反应。2004年,takahiroitoh课题组报道了一种高效的钯催化的硫醇和芳香卤代物形成碳硫键的反应,1,4-二氧六环为溶剂[takahiroi.org.lett.2004,6,4587–4590],但是反应生成的卤代副产物污染环境。2018年,liu课题组报道了一种高效铜催化串联法制备2-芳基噻吩并噻唑的工艺,通过2-氨基苯乙硫醇与四甲基硫代硫化氢(tmtd)缩合,原位生成2-巯基苯并噻唑,并与碘苯偶联,使该方法在有机合成中具有实用价值[liux.j.org.chem.2018,83,11703–11711]。然而,对于活性低的卤芳烃,这些反应往往需要较高的温度和较长的反应时间。2018年,wang课题组报道了一种分子氧为氧化剂,甲苯为溶剂,芳基肼与甲硫醇的钯催化氧化交叉偶联反应。该方法为非对称二芳基硫化物的制备提供了一种高效的方法[wangc,l.j.org.chem.2018,83,2389–2394],但是该方法中肼类底物与硫酚类底物的比例需保持在1:2,反应副产物多,原子经济性差,给后处理带来不便,且反应使用高沸点的有机溶剂,对环境危害大,危废处理也增加了反应成本。
技术实现要素:
本发明针对现有技术中不对称硫化物的制备存在有机溶剂污染、反应副产物多的问题,提供一种水溶性过渡金属配合物催化分子氧氧化水相合成不对称硫化物的方法,本发明利用分子氧作为氧化剂,以水作溶剂,避免了有机溶剂的使用,且收率高,基本不存在副产物的问题。
本发明采用如下技术方案:
一种水溶性过渡金属配合物催化分子氧氧化水相合成不对称硫化物的方法,以摩尔比为1:1的巯基化合物和肼类化合物为底物并将其分散于碱性水溶液中,在40-100℃且氧气存在条件下,以水溶性过渡金属配合物为催化剂,搅拌反应即得不对称硫化物。
优选地,所述肼基化合物具有以下结构通式:
r1-nhnh2
其中r1表示以下取代基:脂肪烷基、环烷基、萘基、杂环基以及有甲基、甲氧基、硝基、氰基、氟基、氯基或溴基取代的苯基。
优选地,所述巯基化合物具有以下结构通式:
r2-sh
其中r2表示以下取代基:脂肪烷基、环烷基、杂环基以及有甲基、甲氧基、硝基、氨基、氯基或溴基取代的苯基。
优选地,所述的不对称硫化物具有以下结构通式:
r1-s-r2。
优选地,所述水溶性过渡金属配合物是由摩尔比为1:1的水溶性配体和金属中心原位配位所得,所述金属中心为pd,cu,fe,ni,mn,co和zn中的任意一种;所述水溶性配体为l1或l2中的一种,所述l1和l2结构如下:
l1:
水溶性配体l1和l2的合成
水溶性配体在查阅相关文献的基础上合成[刘其生.以邻菲啰啉及其衍生物为配体的新型配合物的合成及性质研究[d].山东:山东师范大学,2008.],具体方法如下:
在25mlschlenk中加入85mg的氢化钠(3.6mmol,6eq.)和0.48ml的三甘醇单甲醚(3mmol,5eq.),然后加入2mldmf,在室温条件下敞口搅拌至反应体系中不再有气泡冒出,然后加入150mg的2,9-二氯-1,10-邻菲啰啉150mg(0.6mmol),封管,反应在100℃的油浴中搅拌加热反应4h。反应结束后,将反应管冷却至室温,加入50ml饱和食盐水,用3*50ml二氯甲烷萃取三次,再用饱和食盐水洗涤有机相3-5遍,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。接着以柱层析色谱法将反应混合物分离提纯(展开剂:乙酸乙酯:甲醇=10:1)即可得到目标化合物配体l1,反应收率90%。配体l2在相同的反应条件下合成,反应收率93%。
优选地,所述水溶性过渡金属配合物的用量为巯基化合物或肼类化合物底物摩尔量的0.05%-10%。
优选地,所述碱性水溶液通过向水中加入碱配制得到,所述碱为碳酸铯、碳酸钾、碳酸钠、氢氧化钠、氢氧化钾、氟化钠、氟化钾、氟化铯、dbu和tmeda中任意一种,碱的用量为巯基化合物或肼类化合物底物摩尔量的1-10倍。
优选地,所述氧气存在条件为空气或氧气,当反应气体氛围为氧气时,氧气压为0.1-0.5mpa。
优选地,所述搅拌反应时间为4-24h。
优选地,上述反应结束后,水相母液作为催化剂可重复利用5-10次。
本发明的有益效果如下:
在本发明方法中,利用分子氧作为氧化剂,以水作溶剂,一方面氧气在自然界中广泛存在,廉价易得,还是可再生资源,且氧气的氧化特性较为温和,相比现有技术中常用的有机氧化剂,不会发生过氧化而产生多种副产物的问题,安全可靠,最重要的是,氧气的氧化副产物是水,绿色无污染;另一方面,水作溶剂安全无污染,后处理简单安全,无有机废液产生,可用作大规模工业生产。因此,本发明相较于传统的合成方法,避免了氧化剂和有机溶剂的使用,减少反应废弃物和废液,而副产物主要是水,保证了绿色环保生产。
而且,本发明所用的肼类底物作为环保型试剂,其唯一的副产物是水和氮气,对环境无污染;而水溶性过渡金属配合物催化剂活性高,可多次循环利用,同时避免了贵金属的使用,降低成本;尤为重要的是,本发明的原料投料比为1:1的情况下,反应收率仍能达到99%,符合原子经济的理念,合成工艺简捷,产物选择性高,副产物少,废物少,具有较强的工业应用前景。
附图说明
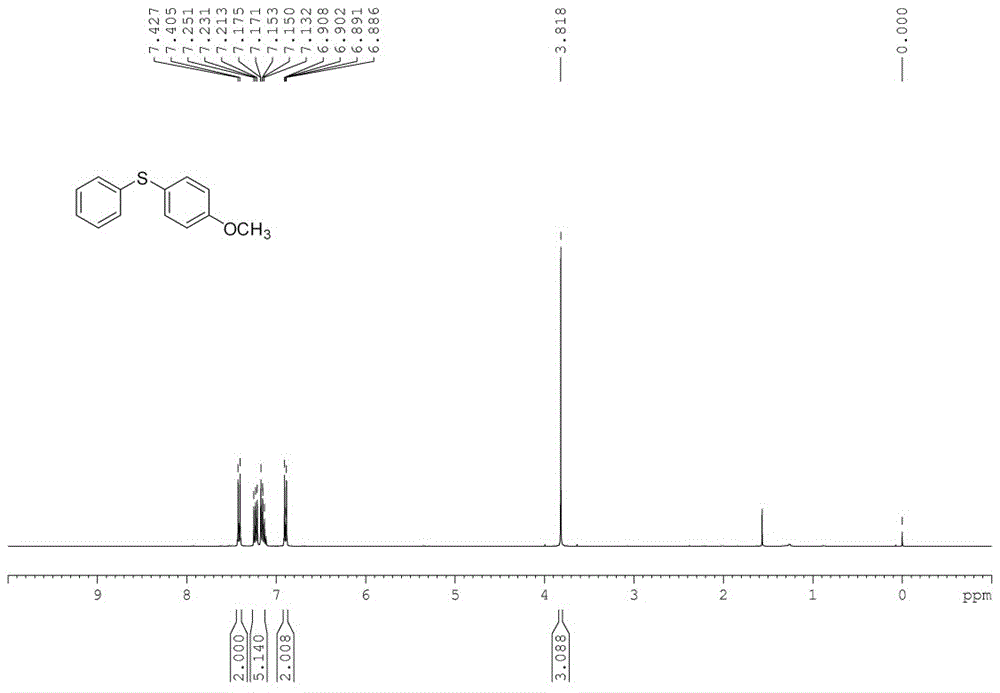
图1是实施例1制备的1-甲氧基-4-(苯硫基)-苯的1hnmr谱图;
图2是实施例1制备的1-甲氧基-4-(苯硫基)-苯的13cnmr谱图。
图3是实施例2制备的4-[(4-甲氧基苯基)硫基]-1,2-二甲基苯的1hnmr谱图;
图4是实施例2制备的4-[(4-甲氧基苯基)硫基]-1,2-二甲基苯的13cnmr谱图。
图5是实施例3制备的1-氯-3-[(4-甲氧基苯基)硫基]-苯的1hnmr谱图;
图6是实施例3制备的1-氯-3-[(4-甲氧基苯基)硫基]-苯的13cnmr谱图。
图7是实施例4制备的1-[(4-甲氧基苯基)硫基]-2-硝基-苯的1hnmr谱图;
图8是实施例4制备的1-[(4-甲氧基苯基)硫基]-2-硝基-苯的13cnmr谱图。
图9是实施例5制备的2-[(4-甲氧基苯基)硫基]-吡啶的1hnmr谱图;
图10是实施例5制备的2-[(4-甲氧基苯基)硫基]-吡啶的13cnmr谱图。
图11是实施例6制备的1-[(1,1-二甲基乙基)硫基]-4-甲氧基苯的1hnmr谱图;
图12是实施例6制备的1-[(1,1-二甲基乙基)硫基]-4-甲氧基苯的13cnmr谱图。
图13是实施例7制备的4-(苯硫基)-苯胺的1hnmr谱图;
图14是实施例7制备的4-(苯硫基)-苯胺的13cnmr谱图。
图15是实施例8制备的2-(苯基硫基)-吡啶的1hnmr谱图;
图16是实施例8制备的2-(苯基硫基)-吡啶的13cnmr谱图。
图17是实施例9制备的2-(苯硫硫基)-苯并噻唑的1hnmr谱图;
图18是实施例9制备的2-(苯硫硫基)-苯并噻唑的13cnmr谱图。
图19是实施例10制备的(己基硫基)-苯的1hnmr谱图;
图20是实施例10制备的(己基硫基)-苯的13cnmr谱图。
具体实施方式
为了使本发明的技术目的、技术方案和有益效果更加清楚,下面结合附图和具体实施例对本发明的技术方案作出进一步的说明。
实施例1
结构式如下的有不对称硫化物的制备方法:
向50ml史莱克管中加入底物苯肼0.2mmol(22mg),对甲氧基苯硫酚0.2mmol(28mg),三水合醋酸铜2.4mg(5mol%,该摩尔量百分比以金属离子摩尔量占底物摩尔量的百分比计算得到,其余实施例的计算方式与此相同),配体l1,5mg(5mol%),碳酸铯65mg(1eq.),水2ml,加入磁子,置换氧气0.1mpa,100℃油浴中搅拌反应12h。反应结束后,将反应管冷却至室温,加入50ml饱和食盐水,用二氯甲烷(3*50ml)萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。接着用柱层析(硅胶柱,30*300mm)法将反应混合物分离提纯(洗脱剂:正己烷:二氯甲烷=2:1),即得到目标产物,收率为99%。目标产物表征数据:yellowoil.(hexane:dcm=2:1aseluent).1hnmr(400mhz,cdcl3)δ=7.43-7.40(m,2h),7.24-7.11(m,5h),6.91-6.87(m,2h),3.82(s,3h).13cnmr(100mhz,cdcl3)δ=159.85,138.64,135.43,128.96,128.18,125.77,124.26,115.01,55.40.lrms(ei)m/zcalcdforc13h12os[m]+,216;found,216。
实施例2
结构式如下的有不对称硫化物的制备方法:
向50ml史莱克管中加入底物3,4-二甲基苯肼0.2mmol(27mg),对甲氧基苯硫酚0.2mmol(28mg),三水合醋酸铜4.8mg(10mol%),配体l2,10mg(10mol%),碳酸铯650mg(10eq.),水2ml,加入磁子,置换氧气0.3mpa,40℃油浴中搅拌反应5h。反应结束后,将反应管冷却至室温,加入50ml饱和食盐水,用二氯甲烷(3*50ml)萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。接着用柱层析(硅胶柱,30*300mm)法将反应混合物分离提纯(洗脱剂:正己烷:二氯甲烷=2:1),即得到目标产物,收率为92%。目标产物表征数据:yellowoil.(hexane:dcm=2:1aseluent).1hnmr(400mhz,cdcl3)δ=7.31-7.25(m,2h),6.99-6.87(m,3h),6.81-6.76(m,2h),3.73(s,3h),2.13(s,3h),2.12(s,3h).13cnmr(100mhz,cdcl3)δ=159.36,137.49,134.98,134.36,134.23,130.69,130.31,127.05,125.82,114.83,55.38,19.79,19.38.lrms(ei)m/zcalcdforc15h16os[m]+,244;found,244。
实施例3
结构式如下的有不对称硫化物的制备方法:
向50ml史莱克管中加入底物3-氯苯肼0.2mmol(28mg),对甲氧基苯硫酚0.2mmol(28mg),二水合氯化铜0.03mg(0.05mol%),配体l2,0.05mg(0.05mol%),碳酸钾84mg(3eq.),水2ml,加入磁子,置换氧气0.2mpa,80℃油浴中搅拌反应10h。反应结束后,将反应管冷却至室温,加入50ml饱和食盐水,用二氯甲烷(3*50ml)萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。接着用柱层析(硅胶柱,30*300mm)法将反应混合物分离提纯(洗脱剂:正己烷:二氯甲烷=2:1),即得到目标产物,收率为98%。目标产物表征数据:yellowsolid.(hexane:dcm=2:1aseluent).50-53℃.1hnmr(400mhz,cdcl3)δ=7.39-7.33(m,2h),7.06(t,j=7.7hz,1h),7.02-6.97(m,2h),6.94-6.90(m,1h),6.87-6.82(m,2h),3.76(s,3h).13cnmr(100mhz,cdcl3)δ=160.34,141.38,136.22,134.81,129.87,126.98,125.59,125.42,122.59,115.25,55.43.lrms(ei)m/zcalcdforc13h11clos[m]+,250;found,250。
实施例4
结构式如下的有不对称硫化物的制备方法:
向50ml史莱克管中加入底物2-硝基苯肼0.2mmol(31mg),对甲氧基苯硫酚0.2mmol(28mg),二水合醋酸锰0.54mg(1mol%),配体l1,1mg(1mol%),碳酸钠176mg(8eq.),水2ml,加入磁子,置换氧气0.5mpa,50℃油浴中搅拌反应8h。反应结束后,将反应管冷却至室温,加入50ml饱和食盐水,用二氯甲烷(3*50ml)萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。接着用柱层析(硅胶柱,30*300mm)法将反应混合物分离提纯(洗脱剂:正己烷:二氯甲烷=2:1),即得到目标产物,收率为99%。目标产物表征数据:yellowsolid.(hexane:dcm=2:1aseluent).91-93℃.1hnmr(400mhz,cdcl3)δ=8.16(d,j=8.2hz,1h),7.43(d,j=8.7hz,2h),7.29-7.22(m,1h),7.11(m,1h),6.93(d,j=8.7hz,2h),6.76(d,j=8.2hz,1h),3.80(s,3h).13cnmr(100mhz,cdcl3)δ=161.20,144.60,140.75,137.77,133.42,127.89,125.80,124.65,121.18,115.71,55.49.lrms(ei)m/zcalcdforc13h11no3s[m]+,261;found,261。
实施例5
结构式如下的有不对称硫化物的制备方法:
向50ml史莱克管中加入底物2-肼吡啶0.2mmol(22mg),对甲氧基苯硫酚0.2mmol(28mg),三氯化铁2.7mg(8mol%),配体l1,8mg(8mol%),tmeda40mg(5eq.),水2ml,加入磁子,置换氧气0.3mpa,70℃油浴中搅拌反应21h。反应结束后,将反应管冷却至室温,加入50ml饱和食盐水,用二氯甲烷(3*50ml)萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。接着用柱层析(硅胶柱,30*300mm)法将反应混合物分离提纯(洗脱剂:正己烷:二氯甲烷=2:1),即得到目标产物,收率为97%。目标产物表征数据:colorlesssolid.(hexane:dcm=2:1aseluent).45-46℃.1hnmr(400mhz,cdcl3)δ=8.33(m,1h),7.49-7.43(m,2h),7.36(m,1h),6.89(m,3h),6.71(d,j=8.1hz,1h),3.78(s,3h).13cnmr(100mhz,cdcl3)δ=162.82,160.70,149.33,137.31,136.74,120.97,120.42,119.48,115.32,55.42.lrms(ei)m/zcalcdforc12h11nos[m]+,217;found,217。
实施例6
结构式如下的有不对称硫化物的制备方法:
向50ml史莱克管中加入底物叔丁基肼0.2mmol(18mg),对甲氧基苯硫酚0.2mmol(28mg),氯化钴0.52mg(2mol%),配体l2,2mg(2mol%),氢氧化钾45mg(4eq.),水2ml,加入磁子,置换氧气0.4mpa,60℃油浴中搅拌反应6h。反应结束后,将反应管冷却至室温,加入50ml饱和食盐水,用二氯甲烷(3*50ml)萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。接着用柱层析(硅胶柱,30*300mm)法将反应混合物分离提纯(洗脱剂:正己烷:二氯甲烷=2:1),即得到目标产物,收率为90%。目标产物表征数据:colorlessoil.(hexane:dcm=2:1aseluent).1hnmr(400mhz,cdcl3)δ=7.40-7.34(m,2h),6.81-6.76(m,2h),3.75(s,3h),1.19(s,9h).13cnmr(100mhz,cdcl3)δ=160.24,138.89,123.62,113.97,55.30,45.52,30.78.lrms(ei)m/zcalcdforc11h16os[m]+,196;found,196。
实施例7
结构式如下的有不对称硫化物的制备方法:
向50ml史莱克管中加入底物苯肼0.2mmol(28mg),对氨基苯硫酚0.2mmol(25mg),氯化镍2.34mg(9mol%),配体l2,9mg(9mol%),氟化钠17mg(2eq.),水2ml,加入磁子,置换氧气0.1mpa,90℃油浴中搅拌反应11h。反应结束后,将反应管冷却至室温,加入50ml饱和食盐水,用二氯甲烷(3*50ml)萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。接着用柱层析(硅胶柱,30*300mm)法将反应混合物分离提纯(洗脱剂:正己烷:二氯甲烷=2:1),即得到目标产物,收率为99%。目标产物表征数据:colorlessoil.(hexane:dcm=2:1aseluent).1hnmr(400mhz,dmso)δ=7.27-7.20(m,2h),7.20-7.14(m,2h),7.13-7.05(m,1h),7.01(m,2h),6.62(d,j=8.6hz,2h),5.53(s,2h).13cnmr(100mhz,dmso)δ=150.05,140.21,136.45,128.97,125.91,124.95,114.82,114.50.lrms(ei)m/zcalcdforc12h11ns[m]+,201;found,201。
实施例8
结构式如下的有不对称硫化物的制备方法:
向50ml史莱克管中加入底物苯肼0.2mmol(28mg),2-巯基吡啶0.2mmol(22.2mg),三水合硝酸铜1.9mg(4mol%),配体l1,4mg(4mol%),氟化钾70mg(6eq.),水2ml,加入磁子,置换氧气0.5mpa,100℃油浴中搅拌反应24h。反应结束后,将反应管冷却至室温,加入50ml饱和食盐水,用二氯甲烷(3*50ml)萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。接着用柱层析(硅胶柱,30*300mm)法将反应混合物分离提纯(洗脱剂:正己烷:乙酸乙酯=10:1),即得到目标产物,收率为96%。目标产物表征数据:brownoil.(hexane:etoac=10:1aseluent).1hnmr(400mhz,cdcl3)δ=8.41-8.30(m,1h),7.57-7.47(m,2h),7.42-7.32(m,4h),6.93(m,1h),6.81(d,j=8.1hz,1h).13cnmr(100mhz,cdcl3)δ=161.57,149.43,136.89,135.02,130.93,129.70,129.19,121.40,119.92.lrms(ei)m/zcalcdforc11h9ns[m]+,187;found,187。
实施例9
结构式如下的有不对称硫化物的制备方法:
向50ml史莱克管中加入底物苯肼0.2mmol(28mg),2-巯基苯并噻唑0.2mmol(33mg),氯化锌1.63mg(6mol%),配体l1,6mg(6mol%),氟化铯760mg(25eq.),水2ml,加入磁子,置换氧气0.1mpa,70℃油浴中搅拌反应7h。反应结束后,将反应管冷却至室温,加入50ml饱和食盐水,用二氯甲烷(3*50ml)萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。接着用柱层析(硅胶柱,30*300mm)法将反应混合物分离提纯(洗脱剂:正己烷),即得到目标产物,收率为97%。目标产物表征数据:yellowoil.(hexaneaseluent).1hnmr(600mhz,cdcl3)δ=7.81(d,j=8.1hz,1h),7.66(d,j=7.2hz,2h),7.57(d,j=7.9hz,1h),7.41(m,3h),7.33(t,j=7.6hz,1h),7.22-7.16(m,1h).13cnmr(150mhz,cdcl3)δ=169.72,153.89,135.56,135.38,130.49,129.99,129.95,126.20,124.37,121.97,120.81.lrms(ei)m/zcalcdforc13h9ns2[m]+,243;found,243。
实施例10
结构式如下的有不对称硫化物的制备方法:
向50ml史莱克管中加入底物苯肼0.2mmol(28mg),正己硫醇0.2mmol(21mg),三水合醋酸铜0.4mg(0.08mol%),配体l2,0.08mg(0.08mol%),碳酸铯260mg(4eq.),水2ml,加入磁子,置换氧气0.3mpa,40℃油浴中搅拌反应15h。反应结束后,将反应管冷却至室温,加入50ml饱和食盐水,用二氯甲烷(3*50ml)萃取三次,加入无水硫酸钠干燥30min,然后利用旋转蒸发仪将低沸点溶剂除去。接着用柱层析(硅胶柱,30*300mm)法将反应混合物分离提纯(洗脱剂:正己烷),即得到目标产物,收率为91%。目标产物表征数据:colorlessoil.(hexaneaseluent).1hnmr(600mhz,cdcl3)δ=7.27-7.12(m,4h),7.05(t,j=7.0hz,1h),2.81(t,j=7.3hz,2h),1.63-1.48(m,2h),1.36-1.15(m,6h),0.79(t,j=6.3hz,3h).13cnmr(150mhz,cdcl3)δ=137.17,128.88,128.84,125.64,33.63,31.44,29.19,28.59,22.61,14.08.lrms(ei)m/zcalcdforc12h8s[m]+,194;found,194。
本发明所适用的不对称硫化物的原料种类、催化剂等均有多种组合,不再以实施例的形式一一表述,现将其中部分组合汇总于下表1中:
表1不同条件下合成的各种结构的不对称硫化物
由以上实施例可知,采用本发明的制备方法,收率均达到了90%以上,该制备方法不需要有机溶剂,反应效率高。
最后所应说明的是:上述实施例仅用于说明而非限制本发明的技术方案,任何对本发明进行的等同替换及不脱离本发明精神和范围的修改或局部替换,其均应涵盖在本发明权利要求保护的范围之内。
- 还没有人留言评论。精彩留言会获得点赞!