一类基于三氮唑结构单元的四齿环金属铂(II)和钯(II)配合物磷光材料的制作方法
本发明涉及磷光发光材料领域,尤其涉及一类含有三氮唑五元杂芳环的四齿环金属铂(ii)和钯(ii)配合物磷光材料。
背景技术:
oled即有机发光二极管(organiclight-emittingdiode)或有机发光器件(organiclight-emittingdevice),又被称为有机电致发光器件(organicelectroluminescentdevice)。有机电致发光是指在顺向偏压电场的作用下有机小分子、金属有机配合物分子或聚合物分子发光材料将电能直接转化为光能的一种发光现象。
oled是自主发光器件,无需背光源,具有响应速度快、驱动电压低、发光效率和分辨率高、对比度高、视角广等特点,此外它能以廉价的玻璃、金属甚至柔性的塑料为基板,因此还具有成本低、生产工艺简单、可进行大面积生产等优点,已成为新一代的全彩显示和照明技术,在手机、电脑、电视、数码相机、gps、可弯曲和折叠等电子产品以及平面固态照明领域具有广阔而巨大的应用前景。
早期器件所采用的发光材料主要为有机小分子荧光材料,而自旋统计量子学表明荧光材料的理论内部量子效率仅为25%。1998年美国普林斯顿大学的forrest教授和南加州大学的thompson教授发现了室温下金属有机配合物分子材料的磷光电致发光现象,利用重金属原子的强自旋轨道耦可以有效促进电子由单线态到三线态的系间蹿越(isc),从而oled器件可以充分利用电激发所产生所有单线态和三线态激发子(exciton),使发光材料的理论内部量子效率可达到100%(nature,1998,395,151)。至此使有机发光材料的研究进入了一个全新的时期。
早期研究的环金属铂(ii)配合物磷光材料多为含有双齿配体和三齿配体的金属有机分子。其刚性较低,两个双齿配体易扭曲、振动而使其磷光量子效率低下(inorg.chem.2002,41,3055);含有三齿配体的环金属铂(ii)配合物由于分子需要第二个配体(如cl-、苯氧基负离子、炔负离子、卡宾等),会使配合物的化学稳定性降低,故双齿和三齿环金属铂(ii)配合物磷光材料均不利于制备稳定而高效的oled器件。而含有四齿配体的环金属铂(ii)配合物分子结构的刚性大大提高,使其磷光量子效率也大为提高,甚至高达100%,且其热稳定性和电化学稳定性亦非常好,因此基于此类磷光材料的oled器件性能也得到了巨大的提高,非常有希望达到了商业化应用的要求。
而对四齿环金属钯(ii)配合物磷光分子的研究尚处于起步阶段,相关报道较少,但是钯盐相对于铂盐更为经济,可降低磷光材料的成本,更为重要的是对于相同配体的铂(ii)配合物磷光分子,钯(ii)配合物磷光分子的发生光谱会发生明显的蓝移,为目前oled领域长期未解决的高效而稳定的蓝光材料的开发提供一个新的途径,对开发新的磷光发光材料具有重要意义。
虽然目前金属有机小分子磷光材料已取得了长足的发展,但是至今为止,在量子效率和稳定性方面皆可满足商业化需求的金属有机小分子还及其有限,因此开发新的磷光材料,尤其是蓝光磷光材料的开发,依然具有举足轻重的意义。
技术实现要素:
本发明针对现有磷光材料技术领域蓝光材料的缺点和不足,尚未实现商业化应用,提供了一类基于含有三唑五元杂芳环的四齿环金属铂(ii)和钯(ii)配合物蓝光磷光材料。
本发明采用如下技术方案:
一种如式1或式2所示的四齿环金属铂(ii)或钯(ii)磷光发光材料:
其中,m1或m2各自独立为铂或钯,r1、r2、r3、r4、r5、r6、r7或r8各自独立为氢、氘、c1-c6的烷基、c1-c6的烷氧基、卤素、胺基、c6-c20的芳基或c6-c20的杂芳基,或杂芳环相邻的两个碳连接相邻位置形成的并环取代基,n1-n8代表取代基的个数,n1或n5各自独立为1~2,n2、n3、n4、n6、n7或n8各自独立为1~3,优选所述的n1-n8各自独立为1,均为最优选n1-n8中任一个均为1。
进一步,所述的四齿环金属铂(ii)磷光发光材料结构式为:
进一步,所述的四齿环金属钯(ii)磷光发光材料结构式为:
本发明所述的各金属配合物磷光材料可以按如下通式方法进行制备,但是不仅限于以下方法:
一种pt1配合物的制备方法,所述方法具体按如下步骤进行:
(1)以1-(3-羟基苯基)-1h-1,2,3-三唑类化合物roh-1、3-溴-(2-吡啶氧基)苯类化合物rbr为原料,碘化亚铜为催化剂,加入配体2-吡啶甲酸和碱磷酸钾,抽换氮气三次,在溶剂二甲基亚砜中,于105℃下搅拌3天,冷却至室温,得到反应混合物a,经大量乙酸乙酯稀释,过滤,乙酸乙酯洗涤,所得滤液用水洗涤3次,无水硫酸钠干燥,过滤,滤液减压旋蒸除去溶剂,将所得粗品通过硅胶柱色谱分离纯化,得目标产物配体a;所述的1-(3-羟基苯基)-1h-1,2,3-三唑类化合物与3-溴-(2-吡啶氧基)苯类化合物、碘化亚铜、2-吡啶甲酸、磷酸钾的物质的量之比为1:1.2~1.5:0.05~0.2:0.1~0.4:2.0~3.0;所述的溶剂的加入量以1-(3-羟基苯基)-1h-1,2,3-三唑类化合物roh-1的物质的量计为1~3ml/mmol;
(2)向所得配体a中加入k2ptcl4和nbu4nbr,抽换氮气三次,然后加入溶剂醋酸在25℃下搅拌12小时后,升温至110℃继续搅拌反应3天,得到反应混合物b,经冷却至室温,减压旋蒸除去溶剂,将所得粗品通过硅胶柱色谱分离纯化,得目标产物pt1配合物;所述配体a与k2ptcl4、nbu4nbr的物质的量配比关系为1:1.0~1.2:0.1~0.2,所述醋酸的加入量以配体a的物质的量计为50~70ml/mmol。
一种pt2配合物的制备方法,所述方法具体按如下步骤进行:
(1)以1-(3-羟基苯基)-2h-1,2,3-三唑类化合物roh-1,3-溴-(2-吡啶氧基)苯类化合物rbr为原料,碘化亚铜为催化剂,加入配体2-吡啶甲酸和碱磷酸钾,抽换氮气三次,在溶剂二甲基亚砜中,105℃下搅拌3天,得到反应混合物c,经冷却至室温,大量乙酸乙酯稀释,过滤,乙酸乙酯洗涤,所得滤液用水洗3次,无水硫酸钠干燥,过滤,滤液减压旋蒸除去溶剂,将所得粗品通过硅胶柱色谱分离纯化,得配体b;所述的1-(3-羟基苯基)-2h-1,2,3-三唑类化合物与3-溴-(2-吡啶氧基)苯类化合物、碘化亚铜、2-吡啶甲酸、磷酸钾的物质的量之比为1:1.2~1.5:0.05~0.2:0.1~0.4:2.0~3.0;所述的溶剂的加入量以1-(3-羟基苯基)-2h-1,2,3-三唑类化合物roh-2的物质的量计为1ml/mmol~3ml/mmol;
(2)向所得配体b中加入k2ptcl4和nbu4nbr,抽换氮气三次,在溶剂醋酸中,于25℃下搅拌12小时后,升温至110℃继续搅拌反应3天,得到反应混合物经冷却至室温,减压旋蒸除去溶剂,将所得粗品通过硅胶柱色谱分离纯化,得目标产物pt2配合物;所述配体b与k2ptcl4、nbu4nbr的物质的量配比关系为1:1.0~1.2:0.1~0.2,所述醋酸的加入量以配体b的物质的量计为50~70ml/mmol。
一种pd1配合物的制备方法,所述方法具体按如下步骤进行:
向所得配体a中加入pd(oac)2和nbu4nbr,抽换氮气三次,在溶剂醋酸中,于110℃下搅拌2天,得到反应混合物冷却至室温,减压蒸馏除去溶剂,将所得粗品通过硅胶柱色谱分离纯化,得目标产物pd1配合物;所述的配体a与pd(oac)2、nbu4nbr的物质的量之比为:1:1.0~1.2:0.1~0.2,所述醋酸的加入量以配体a的物质的量计为50~70ml/mmol。
一种pd1配合物的制备方法,所述方法具体按如下步骤进行:
向所述配体b中加入pd(oac)2和nbu4nbr,抽换氮气三次,在溶剂醋酸中,于110℃下搅拌2天,得到反应混合物冷却至室温,减压蒸馏除去溶剂,将所得粗品通过硅胶柱色谱分离纯化,得目标产物pd2配合物。
本发明所述的四齿环金属铂(ii)或钯(ii)磷光发光材料在作为有机发光器件的发光层中的应用。
更为具体地,本发明所述的四齿环金属铂(ii)或钯(ii)磷光发光材料可作为ito/hatcn/npd/磷光发光材料的有机发光器件主体材料/balq/alq3/lif/al的发光层中的应用;所述的主体材料可以是cbp、trispcz或bebq2。
与现有技术相比,本发明的的有益效果在于:
(1)环金属铂(ii)和钯(ii)配合物易于合成,结构单一、分子量确定,易于纯化,不会产生铱(iii)基配合物中的facial和meridional异构体;
(2)分子刚性强,可以有效减少由于分子振动所消耗的能量,磷光发光强度高;产物可作为一种有机磷光发光材料,可用作有机发光器件的发光层;
(3)相对于双齿和三齿配合物而言,四齿环金属铂(ii)配合物可以有效提高分子的化学稳定性和热稳定性,利于其在oled器件中的应用;
(4)为蓝光磷光材料的开发提供了一个新的途径,具有重要意义。
附图说明
图1为实施例1中有机磷光发光材料pt1在室温下二氯甲烷溶液中的发射光谱图。
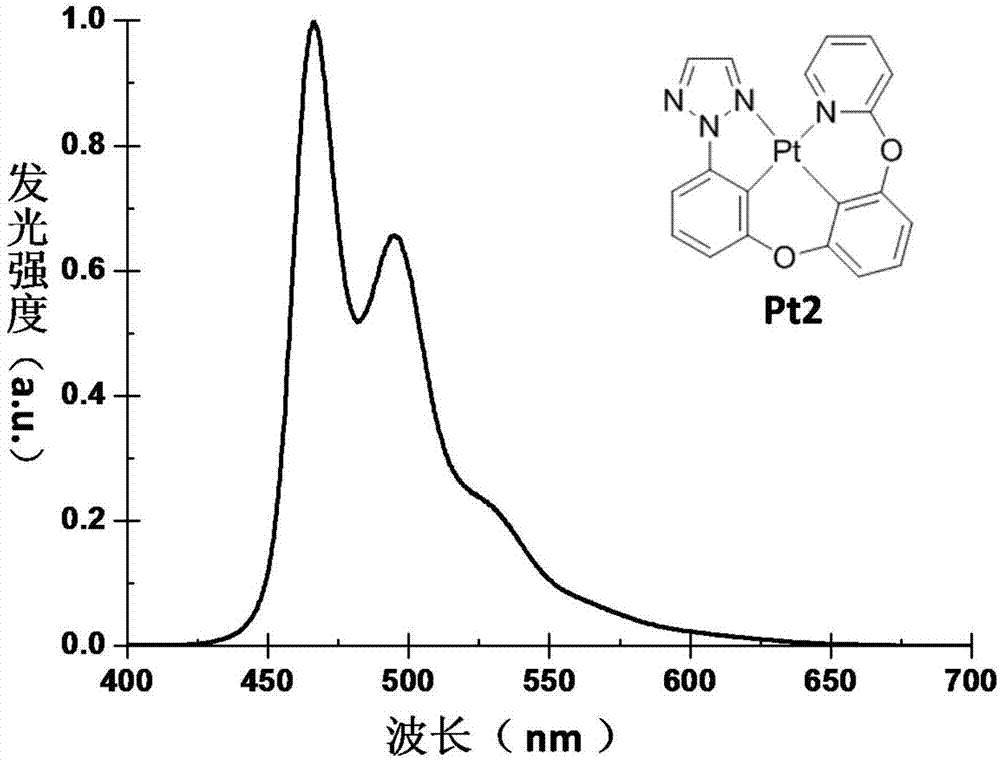
图2为实施例2中有机磷光发光材料pt2在室温下二氯甲烷溶液中的发射光谱图。
图3为实施例3中有机磷光发光材料pd1在室温下二氯甲烷溶液中的发射光谱图。
图4为实施例4中有机磷光发光材料pd2在室温下二氯甲烷溶液中的发射光谱图。
具体实施方式
下面通过具体实施例对本发明作进一步说明,但本发明的保护范围并不仅限于此。
实施例1:四齿环金属铂配合物磷光材料pt1合成路线如下:
3-(3-(1-(1h-1,2,3-三唑基))苯氧基)-(2-吡啶氧基)苯配体1的合成:向带有磁力转子的干燥25ml反应管中依次加入1-(3-羟基苯基)-1h-1,2,3-三唑(644.6mg,4.0mmol,1.0eq),3-溴-(2-吡啶氧基)苯(1.20g,4.8mmol,1.2eq),碘化亚铜(76.2mg,0.4mmol,0.1eq),配体2-吡啶甲酸(98.5mg,0.8mmol,0.2eq),磷酸钾(1.78g,8.4mmol,2.1eq)。抽换氮气三次,然后加入溶剂二甲基亚砜(5ml)。然后反应混合物于105℃下搅拌3天,冷却至室温,大量乙酸乙酯稀释,过滤,乙酸乙酯洗涤。所得滤液用水洗3次,无水硫酸钠干燥。过滤,滤液减压旋蒸除去溶剂,将所得粗品通过硅胶柱色谱分离纯化,淋洗剂(石油醚/乙酸乙酯=3:1-1:1),得目标产物黄色粘稠液体1.00g,收率76%。1hnmr(500mhz,dmso-d6):δ6.90(t,j=2.0hz,1h),6.94-6.97(m,2h),7.06(d,j=8.5hz,1h),7.13-7.17(m,2h),7.46(t,j=8.0hz,1h),7.62(t,j=8.0hz,1h),7.67(t,j=2.0hz,1h),7.73(ddd,j=8.0,2.0,0.5hz,1h),7.86(ddd,j=9.0,7.0,2.0hz,1h),7.98(s,1h),8.15-8.16(m,1h),8.88(d,j=1.0hz,1h).
四齿环金属铂配合物磷光材料pt1的合成:向带有磁力转子和冷凝管的250ml三口瓶中依次加入上步中所得配体1(486.8mg,1.5mmol,1.0eq),k2ptcl4(672.8mg,1.6mmol,1.1eq)和nbu4nbr(48.3mg,0.15mmol,0.1eq)。抽换氮气三次,然后加入溶剂醋酸(88ml)。然后在25℃下搅拌12小时后,升温至110℃继续搅拌反应3天。反应混合物冷却至室温,减压旋蒸除去溶剂,将所得粗品通过硅胶柱色谱分离纯化,淋洗剂(石油醚/二氯甲烷=1:1-1:4),得黄色固体158.5mg,收率20%。1hnmr(500mhz,dmso-d6):δ6.94(dd,j=8.0,1.5hz,1h),7.01(dd,j=8.0,1.0hz,1h),7.06(dd,j=8.0,1.0hz,1h),7.18(t,j=7.5hz,1h),7.32(t,j=8.0hz,1h),7.43-7.46(m,1h),7.51(dd,j=8.5,0.5hz,1h),7.67(dd,j=7.5,0.5hz,1h),8.22(ddd,j=9.0,7.5,2.0hz,1h),8.37(d,j=1.5hz,1h),9.37(d,j=1.0hz,1h),9.71(dd,j=6.0,1.5hz,1h).
制得的pt1在室温下二氯甲烷溶液中的发射光谱图见图1。可见pt1可作为一种有机磷光发光材料,可用作有机发光器件的发光层。
实施例2:四齿环金属铂配合物磷光材料pt2合成路线如下:
3-(3-(2-(2h-1,2,3-三唑基))苯氧基)-(2-吡啶氧基)苯配体2的合成:向带有磁力转子的干燥25ml反应管中依次加入2-(3-羟基苯基)-2h-1,2,3-三唑(644.6mg,4.0mmol,1.0eq),3-溴-(2-吡啶氧基)苯(1.20g,4.8mmol,1.2eq),碘化亚铜(76.2mg,0.4mmol,0.1eq),配体2-吡啶甲酸(98.5mg,0.8mmol,0.2eq),磷酸钾(1.78g,8.4mmol,2.1eq)。抽换氮气三次,然后加入溶剂二甲基亚砜(5ml)。然后反应混合物于105℃下搅拌3天,冷却至室温,大量乙酸乙酯稀释,过滤,乙酸乙酯洗涤。所得滤液用水洗3次,无水硫酸钠干燥。过滤,滤液减压旋蒸除去溶剂,将所得粗品通过硅胶柱色谱分离纯化,淋洗剂(石油醚/乙酸乙酯=10:1-3:1),得目标产物黄色粘稠液体1.20g,收率91%。1hnmr(500mhz,dmso-d6):δ6.92(t,j=2.5hz,1h),6.96-6.99(m,2h),7.06(d,j=8.0hz,1h),7.11-7.15(m,2h),7.46(t,j=8.5hz,1h),7.59(t,j=8.0hz,1h),7.62(t,j=2.0hz,1h),7.81(ddd,j=8.0,2.0,1.0hz,1h),7.86(ddd,j=8.5,7.5,2.0hz,1h),8.13(s,2h),8.17(ddd,j=5.0,2.0,0.5hz,1h).
四齿环金属铂配合物磷光材料pt2的合成(z-9-6):向带有磁力转子和冷凝管的250ml三口瓶中依次加入上步中所得配体2(559.3mg,1.7mmol,1.0eq),k2ptcl4(773.9mg,1.9mmol,1.1eq)和nbu4nbr(54.8mg,0.17mmol,0.1eq)。抽换氮气三次,然后加入溶剂醋酸(102ml)。然后在25℃下搅拌12小时后,升温至110℃继续搅拌反应3天。反应混合物冷却至室温,减压旋蒸除去溶剂,将所得粗品通过硅胶柱色谱分离纯化,淋洗剂(石油醚/二氯甲烷=1:1-1:4),得黄色固体663.9mg,收率75%。1hnmr(500mhz,dmso-d6):δ6.94(dd,j=8.0,1.0hz,1h),7.01(dd,j=8.0,1.0hz,1h),7.05(dd,j=3.0,0.5hz,1h),7.15(t,j=7.5hz,1h),7.28(t,j=8.0hz,1h),7.41-7.44(m,1h),7.49(dd,j=7.5,1.0hz,1h),7.57(dd,j=8.0,1.0hz,1h),8.23-8.26(m,1h),8.42(d,j=0.5hz,1h),8.46(d,j=0.5hz,1h),8.90(dd,j=5.5,1.5hz,1h).
制得的pt2在室温下二氯甲烷溶液中的发射光谱图见图2。可见pt2可作为一种有机磷光发光材料,可用作有机发光器件的发光层。
实施例3:四齿环金属铂配合物磷光材料pd1合成路线如下:
向带有磁力转子和冷凝管的250ml三口瓶中依次加入上步中所得配体1(486.8mg,1.5mmol,1.0eq),pd(oac)2(364.2mg,1.6mmol,1.1eq)和nbu4nbr(48.3mg,0.15mmol,0.1eq)。抽换氮气三次,然后加入溶剂醋酸(88ml)。然后在110℃搅拌反应2天。反应混合物冷却至室温,减压旋蒸除去溶剂,将所得粗品通过硅胶柱色谱分离纯化,淋洗剂(石油醚/二氯甲烷=1:1-1:4),得白色固体417.9mg,收率64%。
1hnmr(500mhz,dmso-d6):δ6.94(dd,j=6.0,1.5hz,1h),7.04(dd,j=8.0,1.0hz,1h),7.11(dd,j=8.0,0.5hz,1h),7.23(t,j=8.0hz,1h),7.36(t,j=8.0hz,1h),7.44-7.48(m,2h),7.68(dd,j=8.0,1.0hz,1h),8.17(ddd,j=9.0,7.5,2.0hz,1h),8.30(d,j=1.5hz,1h),9.34(d,j=1.5hz,1h),9.36(ddd,j=5.5,2.0,0.5hz,1h).
制得的pd1在室温下二氯甲烷溶液中的发射光谱图见图3,可见pd1可作为一种有机磷光发光材料,可用作有机发光器件的发光层。
实施例4:四齿环金属铂配合物磷光材料pd2合成路线如下:
四齿环金属铂配合物磷光材料pd2的合成:向带有磁力转子和冷凝管的250ml三口瓶中依次加入上步中所得配体2(559.3mg,1.7mmol,1.0eq),pd(oac)2(426.6mg,1.9mmol,1.1eq)和nbu4nbr(54.8mg,0.17mmol,0.1eq)。抽换氮气三次,然后加入溶剂醋酸(102ml)。然后在110℃下搅拌2天。反应混合物冷却至室温,减压蒸馏除去溶剂,将所得粗品通过硅胶柱色谱分离纯化,淋洗剂(石油醚/二氯甲烷=1:1-1:4),得白色固体602.4mg,收率82%。1hnmr(500mhz,dmso-d6):δ6.95(dd,j=7.5,1.0hz,1h),7.04(dd,j=8.0,1.0hz,1h),7.09(dd,j=8.5,1.0hz,1h),7.21(t,j=8.0hz,1h),7.32(t,j=8.0hz,1h),7.43-7.46(m,1h),7.51-7.54(m,2h),8.19(ddd,j=8.5,7.0,1.5hz,1h),8.35(d,j=0.5hz,1h),8.42(d,j=0.5hz,1h),8.44(dd,j=5.5,1.5hz,1h).
制得的pd2在室温下二氯甲烷溶液中的发射光谱图见图4,可见pd2可作为一种有机磷光发光材料,可用作有机发光器件的发光层。
- 还没有人留言评论。精彩留言会获得点赞!