白藜芦醇氨基酸酯类衍生物及其制备方法与流程
本发明属于精细化学物质合成技术领域,具体涉及一种白藜芦醇氨基酸酯类衍生物及其制备方法。
背景技术:
白藜芦醇是从1938年被日本科学家从白藜芦中发现的一种天然产物,主要存在于来源于花生、葡萄(红葡萄酒)、虎杖等植物中。现代研究表明白藜芦醇是一种非常重要的食品添加剂和预防医药用品,其对与炎症、心血管疾病、抗疲劳方面有良好的效果。
丙氨酸是组成人体蛋白质的20种氨基酸之一,有助缓和低血糖,改善身体能量状况。γ-氨基丁酸是一种天然存在的非蛋白组成氨基酸,具有极其重要的生理功能,它能促进脑的活化性,健脑益智,抗癫痫,促进睡眠,美容润肤,延缓脑衰老机能,能补充人体抑制性神经递质,具有良好的降血压功效。
现有技术中,往往将白藜芦醇、丙氨酸或γ-氨基丁酸单独使用,用于美容产品或作为抗疲劳、降血压的药物。由于白藜芦醇中活性羟基的存在,使得白藜芦醇在常温下对光、温度、酸碱不稳定,将羟基通过发生化学反应,将其保护起来,可以大大提高白藜芦醇的稳定性。然而,新增加的取代基团往往增加了白藜芦醇的药理毒性,影响白藜芦醇的药用效果。因此,有必要合成一种白藜芦醇的新的衍生物,以降低取代基团所引入的药理毒性,并同时发挥出白藜芦醇维持白藜芦醇的药用效果。
技术实现要素:
有鉴于此,本发明提供了一种新的白藜芦醇的衍生物,即白藜芦醇氨基酸酯类衍生物。
本发明还提供了一种上述白藜芦醇氨基酸酯类衍生物的新的合成方法。
本发明解决其技术问题所采用的技术方案是:
一种白藜芦醇氨基酸酯类衍生物,其结构如式ⅰ所示:
其中,r选自-ch2-ch2-、
一种如上所述的白藜芦醇氨基酸酯类衍生物的制备方法,以白藜芦醇、r氨基酸、r’醇、二氯亚砜、二(对硝基苯)碳酸酯为原料,以4-二甲氨基吡啶(dmap)为催化剂,以乙腈为溶剂,合成白藜芦醇氨基酸酯类衍生物衍生物;
其中,r氨基酸选自α-丙氨酸、β-丙氨酸或γ-氨基丁酸中的一种,r’醇选自甲醇、乙醇或正丙醇的中的一种。
具体地,包括以下步骤:
a.向r’醇中加入二氯亚砜,冰水浴下充分搅拌,加入r氨基酸,在20℃~80℃温度下,充分搅拌6h~12h后,蒸馏除去过量的r’醇,得到固体状的r氨基酸r’酯衍生物;其中,r氨基酸选自α-丙氨酸、β-丙氨酸或γ-氨基丁酸中的一种,r’醇选自甲醇、乙醇或正丙醇的中的一种;
b.将步骤a得到的r氨基酸r’酯衍生物、二(对硝基苯)碳酸酯及dmap加入过量的乙腈溶剂中,其中,二(对硝基苯)碳酸酯的物质的量为r氨基酸r’酯衍生物的1倍~5倍;在20℃~90℃的反应温度下,充分搅拌反应2h~6h,得到中间产物混合溶液;
c.向步骤b得到的中间产物混合溶液中加入盐酸进行洗涤,向洗涤液中加入二氯甲烷进行静置萃取,萃取相依次经干燥、旋蒸及柱层析操作,得到固体状中间产物;
d.将白藜芦醇、步骤c得到的固体状中间产物及dmap加入过量的乙腈溶剂中,其中,步骤c得到的固体状中间产物的物质的量为白藜芦醇的3倍~7倍,在20℃~90℃的反应温度下,充分搅拌反应12h~36h,得到最终产物混合溶液;
e.向步骤d得到的最终产物混合溶液中加入盐酸进行洗涤,向洗涤液中加入二氯甲烷进行静置萃取,萃取相依次经干燥、旋蒸及柱层析操作,得到固体状产物。
本发明采用上述技术方案,其有益效果在于:提供了一种全新的白藜芦醇氨基酸酯类衍生物,将不容易保存的白藜芦醇和丙氨酸或γ-氨基丁酸合成新的化合物,解决了白藜芦醇不易保存的技术问题的同时,降低白藜芦醇取代基团所引入的药理毒性。白藜芦醇结构中具有的羟基与丙氨酸或γ-氨基丁酸中的氨基脱水,以酰胺键的形式相连,当衍生物通过口服的方式进入体内后,形成的化学键可以被体内的酶所水解,释放出白藜芦醇和相应的氨基酸单体,进而发挥各自的生理作用,避免引入其他取代基团而使得白藜芦醇的毒副作用增强。可以预期,该新型化合物能够在美容以及制备抗疲劳、降血压的药物方面发挥重要作用。本发明还提供了一种上述白藜芦醇氨基酸酯类衍生物的制备方法,以白藜芦醇、丙氨酸或γ-氨基丁酸、甲醇/乙醇或正丙醇、二氯亚砜、二(对硝基苯)碳酸酯为原料,以4-二甲氨基吡啶(dmap)为催化剂,以乙腈为溶剂,合成白藜芦醇氨基酸酯类衍生物衍生物,工艺过程简单,白藜芦醇转化率高,产品收率稳定。
附图说明
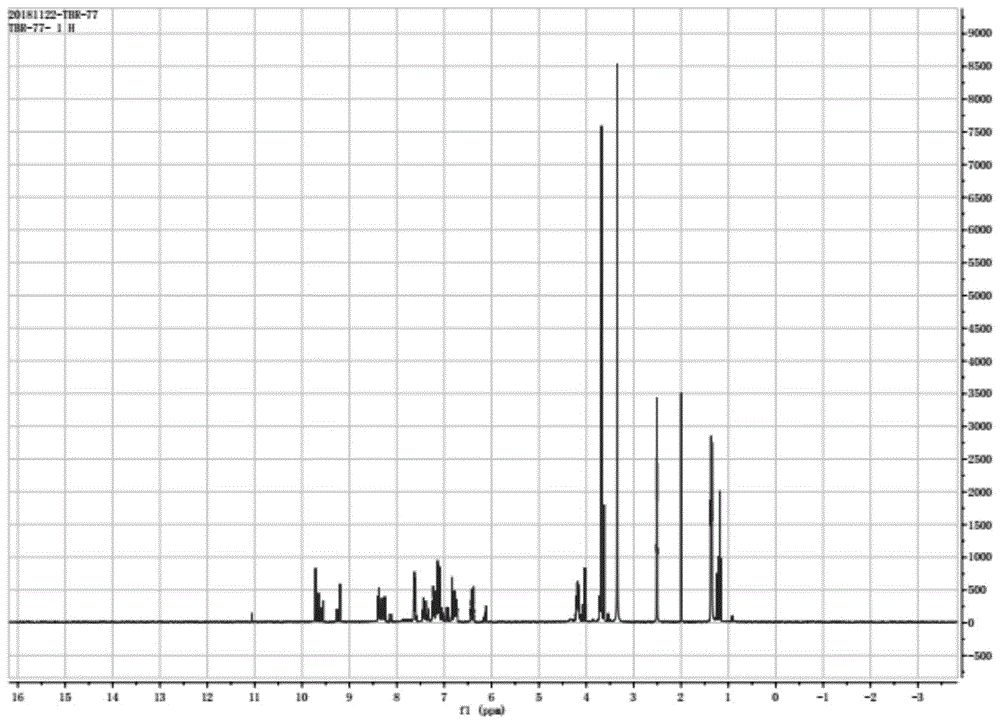
图1是产物p1的核磁氢谱图。
图2是产物p2的核磁氢谱图。
图3是产物p3的核磁氢谱图。
图4是产物p8的核磁氢谱图。
图5是产物p9的核磁氢谱图。
具体实施方式
为了更清楚地说明本发明实施例的技术方案,下面将对本发明方法做进一步说明。
本发明实施例提供了一种白藜芦醇氨基酸酯类衍生物,其结构如式ⅰ所示:
其中,r选自-ch2-ch2-、
作为优选,所述白藜芦醇氨基酸酯类衍生物为白藜芦醇-α-丙氨酸甲酯,其结构如式ⅱ所示:
作为优选,所述白藜芦醇氨基酸酯类衍生物为白藜芦醇-β-丙氨酸甲酯,其结构如式ⅲ所示:
作为优选,所述白藜芦醇氨基酸酯类衍生物为白藜芦醇-γ-氨基丁酸甲酯或白藜芦醇-γ-氨基丁酸乙酯或白藜芦醇-γ-氨基丁酸丙酯,其结构如式ⅳ所示:
其中,r’选自-ch3、-ch2ch3或-ch2ch2ch3。
一较佳实施例中,一种如上所述的白藜芦醇氨基酸酯类衍生物的制备方法,其以白藜芦醇、r氨基酸、r’醇、二氯亚砜、二(对硝基苯)碳酸酯为原料,以4-二甲氨基吡啶(dmap)为催化剂,以乙腈为溶剂,合成白藜芦醇氨基酸酯类衍生物衍生物。其中,r氨基酸选自α-丙氨酸、β-丙氨酸或γ-氨基丁酸中的一种,r’醇选自甲醇、乙醇或正丙醇的中的一种。
具体地,包括以下步骤:
a.向r’醇中加入二氯亚砜,冰水浴下充分搅拌,加入r氨基酸,在20℃~80℃温度下,充分搅拌6h~12h后,蒸馏除去过量的r’醇,得到固体状的r氨基酸r’酯衍生物;其中,r氨基酸选自α-丙氨酸、β-丙氨酸或γ-氨基丁酸中的一种,r’醇选自甲醇、乙醇或正丙醇的中的一种;
其反应过程包括:
a1.二氯亚砜与r’醇取代反应,生成酰氯,如式(1):
a2.酰氯与r氨基酸取代反应,生成r氨基酸r’酯,如式(2):
作为优选,为提高原料利用率,向r’醇缓慢滴加加入二氯亚砜,冰水浴下充分搅拌,以获取醇酰氯。
b.将步骤a得到的r氨基酸r’酯衍生物、二(对硝基苯)碳酸酯及dmap加入过量的乙腈溶剂中,其中,二(对硝基苯)碳酸酯的物质的量为r氨基酸r’酯衍生物的1倍~5倍。在20℃~90℃的反应温度下,充分搅拌反应2h~6h,得到中间产物混合溶液。
其反应过程如式(3)所示:
c.向步骤b得到的中间产物混合溶液中加入盐酸进行洗涤,向洗涤液中加入二氯甲烷进行静置萃取,萃取相依次经干燥、旋蒸及柱层析操作,得到固体状中间产物。
其中,萃取的萃余相经旋蒸操作,脱除溶剂,以回收利用dmap。
其中,选用无水硫酸钠、硅胶、无水氯化钙、蒙脱石、无水氯化钙或氧化铝中的至少一种对萃取相进行干燥操作。
d.将白藜芦醇、步骤c得到的固体状中间产物及dmap加入过量的乙腈溶剂中,其中,步骤c得到的固体状中间产物的物质的量为白藜芦醇的3倍~7倍,在20℃~90℃的反应温度下,充分搅拌反应12h~36h,得到最终产物混合溶液。
优选地,步骤c得到的固体状中间产物的物质的量为白藜芦醇的4倍~6倍。
其反应过程如式(4)所示:
e.向步骤d得到的最终产物混合溶液中加入盐酸进行洗涤,向洗涤液中加入二氯甲烷进行静置萃取,萃取相依次经干燥、旋蒸及柱层析操作,得到固体状产物。
其中,萃取的萃余相经旋蒸操作,脱除溶剂,以回收利用dmap。
其中,选用无水硫酸钠、硅胶、无水氯化钙、蒙脱石、无水氯化钙或氧化铝中的至少一种对萃取相进行干燥操作。
以下通过具体实施方式,进一步对本发明实施例进行解释说明。
实施例一氨基酸酯类衍生物制备
具体实施方式1
在100ml三颈烧瓶中加入40ml的甲醇和3ml的二氯亚砜,将反应容器在冰水浴下开始搅拌,以使反应物充分混合,进而充分生成酰氯,反应2h,得到相应的醇酰氯。然后在其中加入0.01mol(0.8909g)的α-丙氨酸,与80℃温度下,反应6h,得到相应的α-丙氨酸甲酯0.0184mol(1.8975g),产率达到92%。
具体实施方式2
在100ml三颈烧瓶中加入60ml的甲醇和5ml的二氯亚砜,将反应容器在冰水浴下开始搅拌,以使反应物充分混合,进而充分生成酰氯,反应4h,得到相应的醇酰氯。然后在其中加入0.02mol(1.7826g)的α-丙氨酸,与20℃温度下,反应12h,得到相应的α-丙氨酸甲酯0.0194mol(2.0002g),产率达到97%。
具体实施方式3
在100ml三颈烧瓶中加入60ml的甲醇和5ml的二氯亚砜,将反应容器在冰水浴下开始搅拌,以使反应物充分混合,进而充分生成酰氯,反应6h,得到相应的醇酰氯。然后在其中加入0.02mol(1.7823g)的α-丙氨酸,在60℃温度下,反应8h,得到相应的α-丙氨酸甲酯0.0099mol(1.0210g),产率达到99%。
具体实施方式4
在100ml三颈烧瓶中加入60ml的乙醇和5ml的二氯亚砜,将反应容器在冰水浴下开始搅拌,以使反应物充分混合,进而充分生成酰氯,反应4h,得到相应的醇酰氯。然后在其中加入0.02mol(1.7826g)的α-丙氨酸,在60℃温度下,反应8h,得到相应的α-丙氨酸乙酯0.019mol(2.2263g),产率达到95%。
具体实施方式5
在100ml三颈烧瓶中加入60ml的正丙醇和5ml的二氯亚砜,将反应容器在冰水浴下开始搅拌,以使反应物充分混合,进而充分生成酰氯,反应4h,得到相应的醇酰氯。然后在其中加入0.02mol(1.7826g)的α-丙氨酸,在60℃温度下,反应8h,得到相应的α-丙氨酸丙酯0.018mol(2.363g),产率达到90%。
具体实施方式6
在100ml三颈烧瓶中加入40ml的甲醇和3ml的二氯亚砜,将反应容器在冰水浴下开始搅拌,以使反应物充分混合,进而充分生成酰氯,反应2h,得到相应的醇酰氯。然后在其中加入0.01mol(0.89091g)的β-丙氨酸,与80℃温度下,反应6h,得到相应的β-丙氨酸甲酯0.0091mol(0.9385g),产率达到91%。
具体实施方式7
在100ml三颈烧瓶中加入60ml的甲醇和5ml的二氯亚砜,将反应容器在冰水浴下开始搅拌,以使反应物充分混合,进而充分生成酰氯,反应4h,得到相应的醇酰氯。然后在其中加入0.02mol(1.7818g)的β-丙氨酸,与20℃温度下,反应12h,得到相应的β-丙氨酸甲酯0.018mol(1.8565g),产率达到90%。
具体实施方式8
在100ml三颈烧瓶中加入60ml的甲醇和5ml的二氯亚砜,将反应容器在冰水浴下开始搅拌,以使反应物充分混合,进而充分生成酰氯,反应6h,得到相应的醇酰氯。然后在其中加入0.02mol(1.7818g)的β-丙氨酸,在60℃温度下,反应8h,得到相应的β-丙氨酸甲酯0.019mol(1.9596g),产率达到95%。
具体实施方式9
在100ml三颈烧瓶中加入60ml的乙醇和5ml的二氯亚砜,将反应容器在冰水浴下开始搅拌,以使反应物充分混合,进而充分生成酰氯,反应4h,得到相应的醇酰氯。然后在其中加入0.02mol(1.7818g)的β-丙氨酸,在60℃温度下,反应8h,得到相应的β-丙氨酸乙酯0.0192mol(2.2497g),产率达到96%。
具体实施方式10
在100ml三颈烧瓶中加入60ml的正丙醇和5ml的二氯亚砜,将反应容器在冰水浴下开始搅拌,以使反应物充分混合,进而充分生成酰氯,反应4h,得到相应的醇酰氯。然后在其中加入0.02mol(1.7818g)的β-丙氨酸,在60℃温度下,反应8h,得到相应的β-丙氨酸丙酯0.0178mol(2.3351g),产率达到89%。
具体实施方式11
在100ml三颈烧瓶中加入40ml的甲醇和3ml的二氯亚砜,将反应容器在冰水浴下开始搅拌,以使反应物充分混合,进而充分生成酰氯,反应2h,得到相应的醇酰氯。然后在其中加入0.01mol(1.031g)的γ-氨基丁酸,与80℃温度下,反应6h,得到相应的γ-氨基丁酸甲酯0.0092mol(1.0785g),产率达到92%。
具体实施方式12
在100ml三颈烧瓶中加入60ml的甲醇和5ml的二氯亚砜,将反应容器在冰水浴下开始搅拌,以使反应物充分混合,进而充分生成酰氯,反应4h,得到相应的醇酰氯。然后在其中加入0.02mol(2.0621g)的γ-氨基丁酸,与20℃温度下,反应12h,得到相应的γ-氨基丁酸甲酯0.018mol(2.1143g),产率达到90%。
具体实施方式13
在100ml三颈烧瓶中加入60ml的甲醇和5ml的二氯亚砜,将反应容器在冰水浴下开始搅拌,以使反应物充分混合,进而充分生成酰氯,反应6h,得到相应的醇酰氯。然后在其中加入0.02mol(2.0621g)的γ-氨基丁酸,在60℃温度下,反应8h,得到相应的γ-氨基丁酸甲酯0.0194mol(2.2735g),产率达到97%。
具体实施方式14
在100ml三颈烧瓶中加入60ml的乙醇和5ml的二氯亚砜,将反应容器在冰水浴下开始搅拌,以使反应物充分混合,进而充分生成酰氯,反应4h,得到相应的醇酰氯。然后在其中加入0.02mol(2.0621g)的γ-氨基丁酸,在60℃温度下,反应8h,得到相应的γ-氨基丁酸乙酯0.0184mol(2.4146g),产率达到92%。
具体实施方式15
在100ml三颈烧瓶中加入60ml的正丙醇和5ml的二氯亚砜,将反应容器在冰水浴下开始搅拌,以使反应物充分混合,进而充分生成酰氯,反应4h,得到相应的醇酰氯。然后在其中加入0.02mol(2.0621g)的γ-氨基丁酸,在60℃温度下,反应8h,得到相应的γ-氨基丁酸丙酯0.018mol(2.6148g),产率达到90%。
实施例二中间产物的制备
具体实施方式16
向50ml的乙腈中添加α-丙氨酸甲酯0.005mol(0.5162g),二(对硝基苯)碳酸酯0.025mol(7.6073g)及的4-二甲氨基吡啶(dmap)0.3g,在20℃下充分搅拌反应6h。反应完毕后,将反应液体首先经300ml稀盐酸洗涤,然后经300ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到中间产物a0.004mol(1.0734g),产率达到80%。
具体实施方式17
向100ml的乙腈中添加α-丙氨酸甲酯0.01mol(1.0315g),二(对硝基苯)碳酸酯0.01mol(3.0432g)及的4-二甲氨基吡啶(dmap)0.6g,在60℃下充分搅拌反应3.5h。反应完毕后,将反应液体首先经500ml稀盐酸洗涤,然后经500ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到中间产物a0.009mol(2.4143g),产率达到90%。
具体实施方式18
向100ml的乙腈中添加α-丙氨酸甲酯0.01mol(1.0316g),二(对硝基苯)碳酸酯0.03mol(9.1283g)及的4-二甲氨基吡啶(dmap)0.5g,在90℃下充分搅拌反应2h。反应完毕后,将反应液体首先经500ml稀盐酸洗涤,然后经500ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到中间产物a0.0082mol(2.1993g),产率达到82%。
具体实施方式19
向100ml的乙腈中添加α-丙氨酸乙酯0.01mol(1.1721g),二(对硝基苯)碳酸酯0.01mol(3.0432g)及的4-二甲氨基吡啶(dmap)0.6g,在60℃下充分搅拌反应3.5h。反应完毕后,将反应液体首先经500ml稀盐酸洗涤,然后经500ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到中间产物b0.0085mol(2.3993g),产率达到85%。
具体实施方式20
向100ml的乙腈中添加α-丙氨酸丙酯0.01mol(1.3121g),二(对硝基苯)碳酸酯0.01mol(3.0432g)及的4-二甲氨基吡啶(dmap)0.6g,在60℃下充分搅拌反应3.5h。反应完毕后,将反应液体首先经500ml稀盐酸洗涤,然后经500ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到中间产物c0.0082mol(2.4295g),产率达到82%。
具体实施方式21
向100ml的乙腈中添加β-丙氨酸甲酯0.01mol(1.0311g),二(对硝基苯)碳酸酯0.01mol(3.0432g)及的4-二甲氨基吡啶(dmap)0.4g,在60℃下充分搅拌反应3.5h。反应完毕后,将反应液体首先经500ml稀盐酸洗涤,然后经500ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到中间产物d0.0087mol(2.3333g),产率达到87%。
具体实施方式22
向100ml的乙腈中添加β-丙氨酸乙酯0.01mol(1.1721g),二(对硝基苯)碳酸酯0.01mol(3.0433g)及的4-二甲氨基吡啶(dmap)0.5g,在60℃下充分搅拌反应3.5h。反应完毕后,将反应液体首先经500ml稀盐酸洗涤,然后经500ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到中间产物e0.009mol(2.544g),产率达到90%。
具体实施方式23
向100ml的乙腈中添加β-丙氨酸丙酯0.01mol(1.3123g),二(对硝基苯)碳酸酯0.01mol(3.0431g)及的4-二甲氨基吡啶(dmap)0.5g,在60℃下充分搅拌反应3.5h。反应完毕后,将反应液体首先经500ml稀盐酸洗涤,然后经500ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到中间产物f0.0088mol(2.6075g),产率达到88%。
具体实施方式24
向100ml的乙腈中添加γ-氨基丁酸甲酯0.01mol(1.1721g),二(对硝基苯)碳酸酯0.01mol(3.0432g)及的4-二甲氨基吡啶(dmap)0.5g,在60℃下充分搅拌反应3.5h。反应完毕后,将反应液体首先经500ml稀盐酸洗涤,然后经500ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到中间产物g0.0091mol(2.5683g),产率达到91%。
具体实施方式25
向100ml的乙腈中添加γ-氨基丁酸乙酯0.01mol(1.3121g),二(对硝基苯)碳酸酯0.01mol(3.0432g)及的4-二甲氨基吡啶(dmap)0.5g,在60℃下充分搅拌反应3.5h。反应完毕后,将反应液体首先经500ml稀盐酸洗涤,然后经500ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到中间产物h0.0087mol(2.5776g),产率达到87%。
具体实施方式26
向100ml的乙腈中添加γ-氨基丁酸丙酯0.01mol(1.4521g),二(对硝基苯)碳酸酯0.01mol(3.0432g)及的4-二甲氨基吡啶(dmap)0.5g,在60℃下充分搅拌反应3.5h。反应完毕后,将反应液体首先经500ml稀盐酸洗涤,然后经500ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到中间产物j0.008mol(2.2821g),产率达到80%。
实施例三白藜芦醇氨基酸酯类衍生物的制备
具体实施方式27
向50ml的乙腈中添加所得中间产物a0.003mol(0.8053g),白藜芦醇0.001mol(0.2283g)及4-二甲氨基吡啶(dmap)0.5g,在20℃下充分搅拌反应36h,反应完毕后,将反应液体首先经300ml稀盐酸洗涤,然后经300ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到产物p10.00081mol(0.5373g),产率达到81%。产物p1的核磁氢谱图如图1所示。
具体实施方式28
向100ml的乙腈中添加所得中间产物d0.004mol(1.0732g),白藜芦醇0.001mol(0.2281g)及4-二甲氨基吡啶(dmap)0.6g,在60℃下充分搅拌反应24h,反应完毕后,将反应液体首先经500ml稀盐酸洗涤,然后经500ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到产物p20.00083mol(0.5631g),产率达到83%。产物p2的核磁氢谱图如图2所示。
具体实施方式29
向100ml的乙腈中添加所得中间产物g0.006mol(1.6943g),白藜芦醇0.001mol(0.2282g)及4-二甲氨基吡啶(dmap)0.6g,在90℃下充分搅拌反应12h,反应完毕后,将反应液体首先经500ml稀盐酸洗涤,然后经500ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到产物p30.00079mol(0.5231g),产率达到79%。产物p3的核磁氢谱图如图3所示。
具体实施方式30
向100ml的乙腈中添加所得中间产物b0.005mol(1.4113g),白藜芦醇0.001mol(0.2282g)及4-二甲氨基吡啶(dmap)0.5g,在60℃下充分搅拌反应24h,反应完毕后,将反应液体首先经500ml稀盐酸洗涤,然后经500ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到产物p40.0008mol(0.5383g),产率达到80%。
具体实施方式31
向100ml的乙腈中添加所得中间产物c0.005mol(1.4813g),白藜芦醇0.001mol(0.2282g)及4-二甲氨基吡啶(dmap)0.5g,在60℃下充分搅拌反应24h,反应完毕后,将反应液体首先经500ml稀盐酸洗涤,然后经500ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到产物p50.00082mol(0.5856g),产率达到82%。
具体实施方式32
向100ml的乙腈中添加所得中间产物d0.005mol(1.3413g),白藜芦醇0.001mol(0.2282g)及4-二甲氨基吡啶(dmap)0.5g,在60℃下充分搅拌反应24h,反应完毕后,将反应液体首先经500ml稀盐酸洗涤,然后经500ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到产物p20.00075mol(0.4656g),产率达到75%。
具体实施方式33
向100ml的乙腈中添加所得中间产物e0.005mol(1.4115g),白藜芦醇0.001mol(0.2285g)及4-二甲氨基吡啶(dmap)0.5g,在60℃下充分搅拌反应24h,反应完毕后,将反应液体首先经500ml稀盐酸洗涤,然后经500ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到产物p60.00079mol(0.5413g),产率达到79%。
具体实施方式34
向100ml的乙腈中添加所得中间产物f0.005mol(1.4817g),白藜芦醇0.001mol(0.2287g)及4-二甲氨基吡啶(dmap)0.5g,在60℃下充分搅拌反应24h,反应完毕后,将反应液体首先经500ml稀盐酸洗涤,然后经500ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到产物p70.00085mol(0.6573g),产率达到85%。
具体实施方式35
向100ml的乙腈中添加所得中间产物g0.005mol(1.4110g),白藜芦醇0.001mol(0.2282g)及4-二甲氨基吡啶(dmap)0.5g,在60℃下充分搅拌反应24h,反应完毕后,将反应液体首先经500ml稀盐酸洗涤,然后经500ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到产物p30.00077mol(0.5649g),产率达到77%。
具体实施方式36
向100ml的乙腈中添加所得中间产物h0.005mol(1.4813g),白藜芦醇0.001mol(0.2282g)及4-二甲氨基吡啶(dmap)0.5g,在60℃下充分搅拌反应24h,反应完毕后,将反应液体首先经500ml稀盐酸洗涤,然后经500ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到产物p80.00088mol(0.6158g),产率达到88%。产物p8的核磁氢谱图如图4所示。
具体实施方式37
向100ml的乙腈中添加所得中间产物j0.005mol(1.4263g),白藜芦醇0.001mol(0.2282g)及4-二甲氨基吡啶(dmap)0.5g,在60℃下充分搅拌反应24h,反应完毕后,将反应液体首先经500ml稀盐酸洗涤,然后经500ml二氯甲烷萃取,萃取相用无水硫酸钠干燥,并旋蒸除去溶剂,柱层析得到产物p90.0008mol(0.5932g),产率达到80%。产物p9的核磁氢谱图如图5所示。
以上所揭露的仅为本发明较佳实施例而已,当然不能以此来限定本发明之权利范围,本领域普通技术人员可以理解实现上述实施例的全部或部分流程,并依本发明权利要求所作的等同变化,仍属于发明所涵盖的范围。
- 还没有人留言评论。精彩留言会获得点赞!