利用双相酸水解制备环黄芪醇的方法与流程
本发明属于医药化工领域,具体涉及利用双相酸水解制备环黄芪醇的方法。
背景技术:
黄芪甲苷是中药黄芪的主要成分。研究表明,黄芪甲苷具有很好的强心、保肝、抗肿瘤、降血糖等生物活性。黄芪甲苷虽然有很好的生物活性,但由于它相对分子质量和空间位阻较大,使其生物利用度降低,所以不利于药物制剂的开发。环黄芪醇是黄芪甲苷的苷元,在自然界主要存在于黄芪中,属于环菠萝烷型四环三帖类化合物。由于它相对分子质量和空间位阻较黄芪甲苷小,使其相对黄芪甲苷而言具有更好的脂溶性,更容易透过细胞膜而被机体吸收。目前研究表明,环黄芪醇可以激活端粒酶活性从而提高cd3和cd8t细胞的增殖能力来抑制细胞的衰老。
目前报道制备环黄芪醇的方法主要有酶解法、smith降解法、酸水解法。其中酶解法工艺简单,但对反应条件要求比较苛刻,不利于应用性推广。smith降解法条件温和,并且收率较高,但需要氧化-还原-水解-萃取-纯化五步完成,步骤繁琐,成本较高。直接酸水解法操作简单,成本较低,但由于环黄芪醇在剧烈的酸性条件下极易转化为黄芪醇,所以在剧烈的条件下,直接采用酸水解法,收率很低。
技术实现要素:
本发明的目的是解决现有技术中酸水解收率低的问题,从而建立一种操作简单,成本低廉、收率较高的制备环黄芪醇的方法。
以下是黄芪甲苷、黄芪醇、环黄芪醇的结构式:
本发明是通过以下技术方案实现的,利用双相酸水解制备环黄芪醇的方法,其特征在于,包括以黄芪甲苷为原料,与有机相和酸溶液组成的双相酸水溶液混合,反应,后处理,获得环黄芪醇;其中,有机相与水不互溶或微溶,环黄芪醇在有机相中的溶剂度大于水相。
优选地,所述酸溶液的溶质为hcl。
更为优选地,酸溶液的溶剂为水或水与醇的混合溶液。
作为优选,酸溶液的溶剂为水与醇的混合溶液。
更为优选的是,酸溶液的溶剂为水与甲醇的混合溶液。
优选地,所述有机相为三氯甲烷,二氯甲烷或甲苯;更为优选地,所述有机相为三氯甲烷,经过实验发现当有机相为三氯甲烷时,其收率较高;有机相为该三种的任意一种时,其收率明显较高。
具体地,所述反应的温度为室温,所述反应的时间为2-6天。
优选地,所述黄芪甲苷与酸溶液、有机相的质量体积比为1mg:1ml:1.6ml。
优选地,所述酸溶液的浓度为6%-18%;当酸溶液为酸水和醇的混合溶液时,酸溶液的浓度为6-18%,醇溶液的浓度为5%-20%。
具体地,所述后处理为碳酸氢钠中和水相,收集有机相,氯化钠水洗,无水硫酸钠干燥有机相,减压蒸馏有机相,得到固体产品。
作为优选,所述后处理还包括,将固体产品用三氯甲烷:甲醇体积比为15:1进行洗脱,收集洗脱剂,减压蒸馏,得到提纯的环黄芪醇。
具体地,将黄芪甲苷加入酸溶液中,再加入有机相,反应,后处理,获得环黄芪醇。
有益效果
本发明以黄芪甲苷为原料,是利用双相酸水解的方法制备环黄芪醇,使酸水解生成的环黄芪醇分布在有机相中,避免了环黄芪醇在剧烈的酸水相中下转化为黄芪醇的缺点,有效保护了环黄芪醇的稳定性,大大提高了环黄芪醇的收率,其收率最高可达46.28%,且步骤简单,成本低,适于工业生产;通过静置、分层、萃取、硅胶柱层析分离得到高纯度环黄芪醇,纯度可高达99%。
附图说明
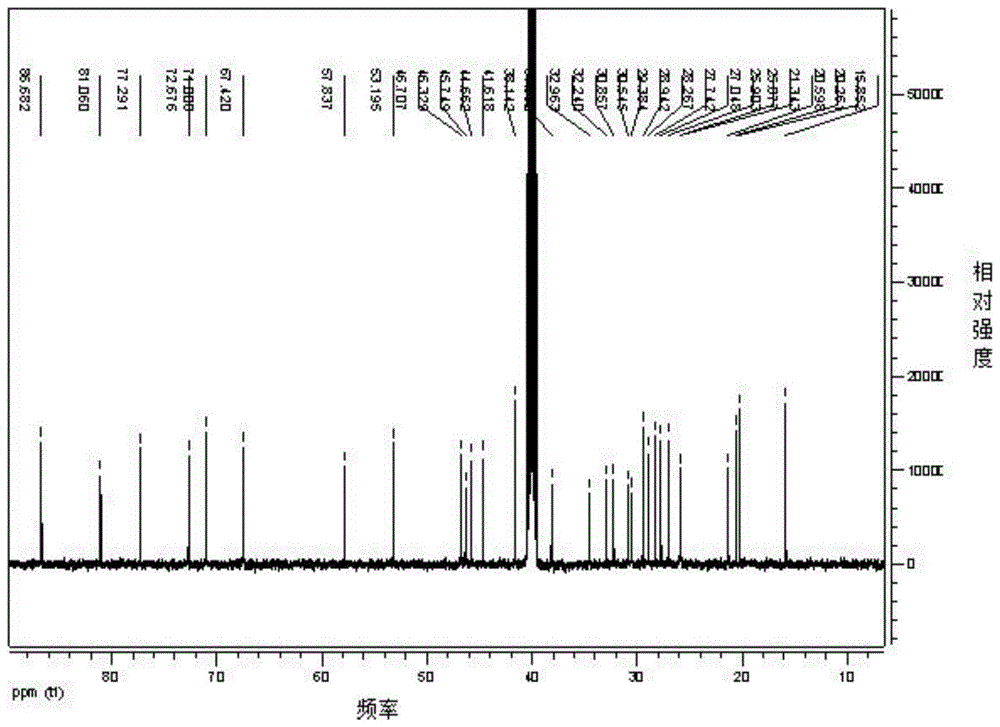
图1实施例1制备的环黄芪醇的核磁c谱图;
图2实施例1制备的环黄芪醇的核磁h谱图;
图3实施例1制备的环黄芪醇的高分辨质谱图。
具体实施方式
实施例1
利用双相酸水解制备环黄芪醇的方法,包括:
双相酸水解制备环黄芪醇:
取400mg黄芪甲苷作为原料,加入到400ml体积分数为18%盐酸的20%甲醇水溶液中,加入有机相三氯甲烷640ml,在室温条件下剧烈搅拌6天,反应后静置,收集有机相三氯甲烷,将酸水相中加入碳酸氢钠中和,再加入等体积三氯甲烷萃取,合并有机相,用饱和氯化钠水溶液清洗,再用无水硫酸钠干燥,减压浓缩,得到粗品。通过hplc检测,计算最终环黄芪醇收率为46.28%。
环黄芪醇的分离:
取200mg上述粗品加入600mg的200-300目硅胶拌样,取8000mg硅胶,用三氯甲烷湿法填柱,将拌有硅胶的粗品均匀散在硅胶柱顶部,然后用三氯甲烷:甲醇体积比为15:1进行洗脱,流速大约为1.0ml/min。每10ml取样一次用薄层色谱进行检测。收集洗脱剂,减压蒸馏得到产品,为纯度99%的环黄芪醇。
将制备所得的产品进行核磁h谱测定,如图2所示,环黄芪醇核磁h谱数据分析:
1hnmr(dmso-d6,400mhz):δ4.87(s,1h),4.67(d,j=2hz,1h),4.48(m,1h),4.21(d,j=3.2hz,1h),4.00(d,j=3.6hz,1h),3.62(t,j=9.2hz,1h),3.29(m,1h),3.03(m,1h),2.54(t,j=14hz,1h),2.18(d,j=5.2hz,1h),1.91(m,2h),1.86(m,1h),1.80(m,1h),1.70(m,1h),1.57(m,2h),1.51(m,2h),1.45(m,2h),1.36(m,1h),1.26(m,2h),1.21(d,j=6.4hz2h),1.18(s,4h),1.13(s,6h),1.11(s,3h),1.02(s,3h),0.89(s,3h),0.82(s,3h),0.44(d,j=2.4hz,1h),0.24(d,j=2.8hz,1h)。
将制备所得的产品进行核磁c谱测定,如图1所示,环黄芪醇核磁c谱数据分析:13cnmr(dmso-d6,100mhz)δ(ppm)86.68,81.06,77.29,72.67,71.00,67.42,57.83,53.19,46.70,46.32,45.74,44.66,41.61,38.14,34.52,32.96,32.23,30.85,30.54,29.38,28.94,28.26,27.74,27.04,25.90,25.87,21.34,20.59,20.26,15.85。
将制备所得的产品进行高分辨质谱分析,如图3所示,hrms:理论值为c30h50o5[m+na]+513.3550,实际测量值为513.3535,其为环黄芪醇。
实施例2
利用双相酸水解制备环黄芪醇的方法,包括:
双相酸水解制备环黄芪醇:
取400mg黄芪甲苷作为原料,加入到400ml体积分数为18%的盐酸20%甲醇水溶液中,加入有机相二氯甲烷640ml,在室温条件下剧烈搅拌6天,反应后静置,收集有机相二氯甲烷,将酸水相加入碳酸氢钠中和,再加入等体积三氯甲烷萃取,合并有机相三氯甲烷和二氯甲烷,减压浓缩,得到粗品。通过hplc检测,计算最终环黄芪醇收率为33.8%。
环黄芪醇的分离:
取200mg上述粗品加入600mg的200-300目硅胶拌样,取8000mg硅胶,用三氯甲烷湿法填柱,将拌有硅胶的粗品均匀散在硅胶柱顶部,然后用三氯甲烷:甲醇体积比为15:1进行洗脱,流速大约为1.0ml/min。每10ml取样一次用薄层色谱进行检测。收集洗脱剂,减压蒸馏得到纯度为98%的环黄芪醇。
实施例3
利用双相酸水解制备环黄芪醇的方法,包括:
双相酸水解制备环黄芪醇:
取400mg黄芪甲苷作为原料,加入到400ml体积分数为6%的盐酸5%甲醇水溶液中,加入有机相三氯甲烷640ml,在室温条件下剧烈搅拌2天,反应后静置,收集有机相三氯甲烷,将酸水相加入碳酸氢钠中和,再加入等体积三氯甲烷萃取,合并有机相,减压浓缩,得到粗品。通过hplc检测,计算最终环黄芪醇收率为18.89%。
环黄芪醇的分离:
取200mg上述粗品加入600mg的200-300目硅胶拌样,取8000mg硅胶,用三氯甲烷湿法填柱,将拌有硅胶的粗品均匀散在硅胶柱顶部,然后用三氯甲烷:甲醇体积比为15:1进行洗脱,流速大约为1.0ml/min。每10ml取样一次用薄层色谱进行检测。收集洗脱剂,减压蒸馏得到纯度为97%的环黄芪醇。
实施例4
利用双相酸水解制备环黄芪醇的方法,包括:
双相酸水解制备环黄芪醇:
取400mg黄芪甲苷作为原料,加入到400ml体积分数为12%盐酸的10%甲醇水溶液中,加入有机相三氯甲烷640ml,在室温条件下剧烈搅拌4天,反应后静置,收集有机相三氯甲烷,将酸水相加入碳酸氢钠中和,再加入等体积三氯甲烷萃取,合并有机相,减压浓缩,得到粗品。通过hplc检测,计算最终环黄芪醇收率为31.78%。
环黄芪醇的分离:
取200mg上述粗品加入600mg的200-300目硅胶拌样,取8000mg硅胶,用三氯甲烷湿法填柱,将拌有硅胶的粗品均匀散在硅胶柱顶部,然后用三氯甲烷:甲醇体积比为15:1进行洗脱,流速大约为1.0ml/min。每10ml取样一次用薄层色谱进行检测。收集洗脱剂,减压蒸馏得到纯度为99%的环黄芪醇。
实施例5
利用双相酸水解制备环黄芪醇的方法,包括:
双相酸水解制备环黄芪醇:
取400mg黄芪甲苷作为原料,加入到400ml体积分数为18%盐酸的20%甲醇水溶液中,加入有机相甲苯640ml,在室温条件下剧烈搅拌6天,反应后静置,收集有机相甲苯,将酸水相加入碳酸氢钠中和,再加入等体积三氯甲烷萃取,合并有机相三氯甲烷和甲苯,减压浓缩,得到粗品。通过hplc检测,计算最终环黄芪醇收率为20.6%。
环黄芪醇的分离:
取200mg上述粗品加入600mg的200-300目硅胶拌样,取8000mg硅胶,用三氯甲烷湿法填柱,将拌有硅胶的粗品均匀散在硅胶柱顶部,然后用三氯甲烷:甲醇体积比为15:1进行洗脱,流速大约为1.0ml/min。每10ml取样一次用薄层色谱进行检测。收集洗脱剂,减压蒸馏得到纯度为98%的环黄芪醇。
对比例1
单相酸水解制备环黄芪醇:
取200mg黄芪甲苷作为原料,加入到200ml体积分数为20%硫酸的20%甲醇水溶液中,加热回流10小时,冷却后,向反应液中加入碳酸氢钠中和,再加入等体积三氯甲烷萃取三次,合并有机相,减压浓缩,得到粗品。通过hplc检测,计算最终环黄芪醇收率为9.6%。
对比例2
单相酸水解制备环黄芪醇:
取200mg黄芪甲苷作为原料,加入到200ml体积分数为10%盐酸的10%甲醇水溶液中,加热回流2小时,冷却后,向反应液中加入碳酸氢钠中和,再加入等体积三氯甲烷萃取三次,合并有机相,减压浓缩,得到粗品。通过hplc检测,计算最终环黄芪醇收率为8.7%。
对比例3
单相酸水解制备环黄芪醇:
取200mg黄芪甲苷作为原料,加入到200ml体积分数为10%盐酸的10%甲醇水溶液中,室温搅拌7天,向反应液中加入碳酸氢钠中和,再加入等体积三氯甲烷萃取三次,合并有机相,减压浓缩,得到粗品。通过hplc检测,计算最终环黄芪醇收率为13.7%。
通过对比例1-3可以看出,单相酸水解制备环黄芪醇,当采用盐酸的甲醇溶液作为水解溶液时,其收率最高可达13.7%,显然远远低于本申请的收率。本发明通过双相酸水解法制备环黄芪醇,在室温条件下,以黄芪甲苷为原料,在盐酸的酸水溶液与有机溶剂形成的两相介质中进行水解,目标产物收率显著提高。
- 还没有人留言评论。精彩留言会获得点赞!