一种基于RNA-Seq开发的小扁豆KASP标记及应用的制作方法
本发明涉及一种基于rna-seq开发的小扁豆kasp标记及应用,涉及分子标记技术领域。
背景技术:
小扁豆是一年生自花受粉、长日性草本植物,同时也是一种重要的冷季食用豆,二倍体(2n=2x=14)植物,基因组大小约为4g。小扁豆是世界第六大食用豆。小扁豆是人类饮食中蛋白质、糖类、维他命和微量元素的重要来源,同时也是牲畜高价值饲料。小扁豆因其固氮能力而有益于作物轮作。但是,由于小扁豆常被种植在贫瘠的土壤中,并且受到干旱、高温胁迫以及各种病害,诸如枯萎病、炭疽病、锈病、颈腐病、根腐病和白霜病等原因的影响,小扁豆的单产量仍然较低。根据fao数据,世界平均单产只有1,152kg/ha。因此,利用合适的育种策略提高小扁豆的产量显得十分迫切。
kasp标记技术(即竞争性等位基因特异性pcr)是目前国际上主流的基因分型方法之一,基于引物末端碱基的特异匹配来对snp和特定位点上的indel进行精准的双等位基因判断。引物采用3条特异性引物,其中上游引物2条,3′端为等位变异碱基,5′端添加通用荧光接头序列,下游引物为普通引物,常规pcr扩增,终点法荧光信号检测。该技术准确率高,灵活性超强,无需昂贵的双色标记探针,最低的dna样本需求,无需全基因组扩增,快速高效,可以高通量检测大量样本的少数几个标记。
目前,kasp标记在小麦、玉米、大豆和花生等作物应用十分广泛。遗憾的是,在小扁豆中没有任何报道。
技术实现要素:
本发明克服了上述现有技术的不足,提供一种基于rna-seq开发的小扁豆kasp标记及应用。本发明通过kasp技术大规模开发了78个小扁豆kasp标记,并且阐述了这些标记在小扁豆种质资源遗传多样性分析方面的应用。
一组基于rna-seq开发的小扁豆kasp标记,包括如下78个kasp标记所对应的正向引物1、正向引物2和反向引物:
本发明还提供了所述小扁豆的kasp标记在小扁豆种质遗传多样性分析中的应用。
进一步的,上述小扁豆的kasp标记的引物通过如下步骤获得:
1)选取6份来源地不同的小扁豆材料,提取rna建立cdna文库,并在illumina平台上测序,获得原始reads;
2)将上述步骤1)获得的原始reads通过数据处理获得cleanreads,将cleanreads组装拼接后形成transcript和unigene;
3)利用步骤2)获得的unigene,依据过滤标准检测出snp位点,选用检测的snp位点设计kasp引物组;
4)提取步骤1)中所述的6份小扁豆材料和另外的选取的88份小扁豆材料(共计94份)的dna作为pcr扩增的dna模板,步骤3)中获得的每组kasp引物构成一个primermix,加入荧光标签后进行pcr扩增,利用带有荧光标签的扩增产物进行基因分型从而验证步骤3)中设计的kasp引物。
进一步的,步骤1)中所述的6份小扁豆材料为小扁豆苗期的整个植株。
进一步的,步骤2)中所述的数据处理包括如下步骤:删除包含adapter的reads、包含ploy-n的reads和低质量reads。
进一步的,步骤3)中所述的过滤标准为:集群:3;windowsize:35;qd<2.0或fs>60.0;mq<40.0或sor>4.0;mqranksum<-12.5或readposranksum<-8.0或dp<10。
进一步的,步骤3)中所述的kasp引物组包括两条特异性正向引物和一条通用反向引物。
进一步的,步骤4)中所述的pcr扩增的反应体系:总体积为4μl:dna模板、2.0μl2×mastermix、0.064μlprimermix、1.936μlddh2o。
进一步的,步骤4)中所述的pcr扩增的反应程序:94℃5min;94℃20s,61℃1min,10循环;94℃20s,55℃1min,26循环;72℃1min。
有益效果:
(1)本发明kasp标记的转化率较高,可以达到60%以上,与snp关联的性状可以很容易地转化为kasp标记,为将来用于小扁豆分子标记辅助育种提供支撑。
(2)kasp标记检测更方便,检测时间短、成本低。在96孔板上可以同时对多个体进行基因分型,增加了检测到稀有杂种个体的可能性。
(3)kasp标记更加灵活,可以通过在引物序列中加入简并或混合碱基来覆盖包含多个snp的序列。
(4)kasp标记检测是定量的,因此可以进行统计分析。
(5)本发明首次设计127个kasp标记对94份小扁豆种质进行基因分型,结果表明78(61.42%)个kasp标记是可用的,其中76(59.84%)个标记具有多态性,2(1.57%)个是单态的,其余标记检测失败。这76个kasp多态标记给小扁豆分子辅助育种提供一种全新的思路。
附图说明
图1kasp引物wd_snp1基于sequencedetectionsystemsv2.4的检测结果。
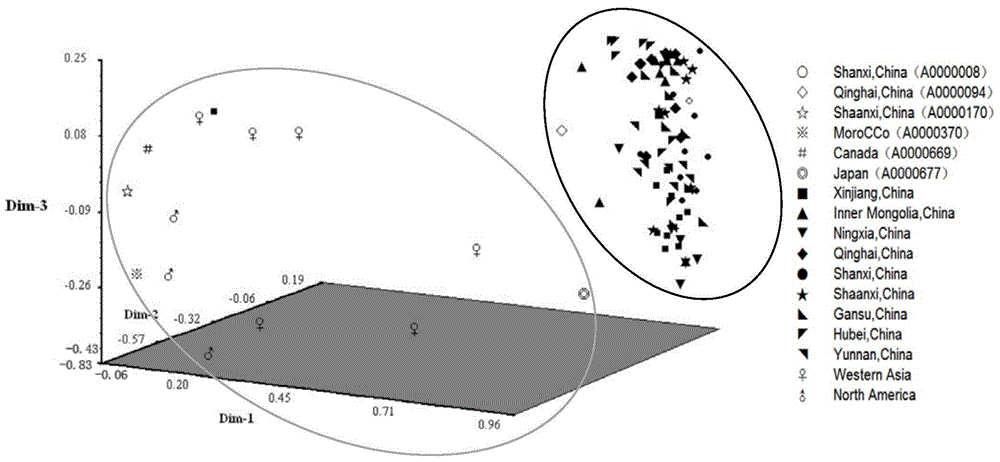
图2基于kasp标记的三维pca分析图。
图3基于kasp标记的upgma系统树图。
图4基于kasp标记的群体结构分析图。
具体实施方式
一、材料与方法
1、植物材料
选取6份小扁豆(lensculinarismedikus)作为试验材料,分别为a0000008来自中国山西,a0000094来自中国青海,a0000170来自中国陕西,a0000370来自摩洛哥,a0000669来自加拿大,a0000677来自日本。每份材料重复三次取样,并标记为a0000008-1,a0000008-2,a0000008-3,a0000094-1,a0000094-2,a0000094-3,a0000170-1,a0000170-2,a0000170-3,a0000370-1,a0000370-2,a0000370-3,a0000669-1,a0000669-2,a0000669-3,a0000677-1,a0000677-2和a0000677-3,同时利用illuminahiseq进行rna-seq。
进行rna-seq的6份小扁豆种质和另外的88份小扁豆种质同时用于kasp检测分析。另外的88份种质来源于3个大的地理区域:79份来自东亚的中国(其中10份来自新疆,9份来自内蒙古,7份来自宁夏,9份来自青海,10份来自山西,7份来自陕西,9份来自甘肃,9份来自湖北,9份来自云南);6份来自西亚,(其中3份来自土耳其,3份来自叙利亚);3份来自北美,(其中2份来自加拿大,1份来自美国)。
实验样品来自于山东省农作物种质资源中心,中国济南。详细信息在表1中给出。
表1.94份小扁豆种质来源地.
2、试验方法
(1)小扁豆illumina测序和转录组组装
利用trizol提取法(tiangen,中国北京)对6份小扁豆苗期(播种后4周)整株(包括根,茎和叶)材料的rna进行分别提取。每个样品取1.5μgrna作为rna样品制备材料。
利用nebnextultrarnalibraryprepkitforillumina(neb,usa)制备测序文库,并将索引代码添加到每个样品的属性序列中。简言之,利用oligo-dt连接的磁珠从总rna中纯化mrna。高温条件下在nebnext第一链合成反应缓冲液(5x)中利用二价阳离子进行破碎,利用随机六聚体引物和m-mulvreversetranscriptase(rnaseh-)合成第一链cdna,随后利用dna聚合酶i和rna酶h合成第二链cdna,通过核酸外切酶/聚合酶活性将剩余的突出末端转化为平末端,将dna片段的3'末端腺苷酸化后,连接具有发夹环结构的nebnext衔接子以准备杂交。,利用ampurexp系统(beckmancoulter,beverly,usa)纯化文库片段从而选择长度为150~200bp的cdna片段,使用3μl的user酶(neb,usa)与大小选择合适、有接头连接的cdna片段在37℃下下共同处理15min,然后在pcr之前95℃下灭活5min。然后以上述处理后的dna片段为模板,用phusionhigh-fidelitydna聚合酶、通用pcr引物和index(x)primer进行pcr。最后,在ampurexp系统中纯化pcr产物,并在agilentbioanalyzer2100系统上评估文库质量。随后,利用truseqpeclusterkitv3-cbot-hs(illumia)在cbotclustergeneration系统上进行索引编码样本的聚类。在簇生成后,制备的文库在illuminahiseq平台上进行测序,并产生双末端reads(即原始reads)。原始数据按顺序存储读取存档(sra),并在ncbi中标记为sub5351043。
上述获得的fastq格式的原始数据(原始reads)首先通过内部perl脚本进行处理。在此步骤中,通过从原始数据中删除包含adapter的reads、包含ploy-n的reads(未知碱基比例大于10%)和低质量reads(q<=20的碱基数占整个read50%以上的reads)来获得cleandata。同时计算了cleandata的q20、q30、gc-含量和序列重复水平。所有的下游分析都是基于高质量的清洁数据。
基于left.fq和right.fq,利用trinity将min_kmer_cov设置为2,而所有其他参数设置为默认值的情况下进行转录组组装。数据去冗余可通过corset(https://code.google.com/p/corset-project/)。corset可以利用比对上transcript的reads数和表达模式对transcript进行层次聚类形成簇,将每个簇中最长的一条transcript保留,称为unigene,并以此进行后续的注释等分析。
为获得全面的基因功能信息,基于以下七大数据库对unigene的功能进行注释:ncbinon-redundantproteinsequences(nr,usingdiamondv0.8.22withe-value=1e-5);ncbinucleotidesequences(nt,usingncbiblast2.2.28+withe-value=1e-5);proteinfamily(pfam,usinghmmer3.0packageandhmmscanwithe-value=0.01);eukaryoticorthologgroups(kog,usingdiamondv0.8.22withe-value=1e-3);swissprot(usingdiamondv0.8.22withe-value=1e-5);kyotoencyclopediaofgenesandgenomes(ko,usingkeggautomaticannotationserverwithe-value=1e-10);geneontology(go,usingblast2gov2.5andself-writingscriptwithe-value=1e-6)。
(2)小扁豆dna提取
利用天根生化科技(北京)公司新型植物基因组dna提取试剂盒(dp320-03),对94份材料(包括rna-seq的6份)的幼苗鲜叶(播种后4周)进行基因组dna的提取,并严格按照厂家的指示进行实验室处理。用nanodropone检测dna质量和浓度,并将dna稀释至25ng/μl储存在-20℃以备pcr所用。
(3)snp搜寻
picard-toolsv1.41和samtoolsv0.1.18用于排序、删除重复读取和合并每个示例的bam对齐结果。使用gatk3软件进行snp调用。使用gatk标准过滤方法和其他参数对原始vcf文件进行过滤(集群:3;windowsize:35;qd<2.0或fs>60.0;mq<40.0或sor>4.0;mqranksum<-12.5或readposranksum<-8.0或dp<10)。
(4)kasp引物设计及检测
根据开发商推荐的标准进行kasp引物设计。两条特异性正向引物3′末端碱基为snp位点变异碱基,5′端分别加上荧光标签fam(gaaggtgaccaagttcatgct)和hex(gaaggtcggagtcaacggatt)序列,同时还有一条通用反向引物,三条引物构成primermix。384孔模块的kasp基本反应体系为4μl:dna模板、2.0μl2×mastermix、0.064μlprimermix、1.936μlddh2o。kasp的pcr反应程序为:94℃5min;94℃20s,61℃1min,10循环;94℃20s,55℃1min,26循环;72℃1min。荧光标签fam和hex用于基因分型。实验所用仪器主要有abi7900ht荧光定量pcr仪(abi7900fast)和9700型双384孔pcr仪(geneamppcrsystem9700)。荧光引物由京梓熙生物科技有限公司合成。sequencedetectionsystemsv2.4用于检测kasp标记结果。
二、数据统计与分析
根据genemapperv4.0分析结果,读取各个荧光标记每个等位基因的数值,低于500的被认为是无效峰,舍弃不用。记录每个等位基因的大小,存储成excel表格,再根据不同软件的格式要求作相应转换。利用ntsys-pcv2.10e进行pca分析。利用powermarkerv3.25计算每个kasp标记的遗传多样性参数,包括:主要等位基因频率、等位基因数、基因多样性、杂合度和多态信息含量(pic)等。结合megav6.0分析软件对参试材料进行基于upgma法的系统发育树构建。利用structurev2.3.4对参试材料进行群体结构分析,参数设定如下:lengthofburninperiod=10000,numberofmcmcrepsafterburnin=10000,亚群数量k(numberofpopulation)=1-20,循环数(numberofiterations)=20。根据deltak(δk)值确定最佳群体结构和亚群数(在线分析的网址为http://taylor0.biology.ucla.edu/struct_harvest/)。
三、结果
(1)小扁豆illumina测序及转录组组装
18个小扁豆样品a0000008-1,a0000008-2,a0000008-3,a0000094-1,a0000094-2,a0000094-3,a0000170-1,a0000170-2,a0000170-3,a0000370-1,a0000370-2,a0000370-3,a0000669-1,a0000669-2,a0000669-3,a0000677-1,a0000677-2和a0000677-3通过illuminahiseq系统测序分别产生了0.677、0.713、0.746、0.684、0.816、0.755、0.888、0.890、0.749、0.810、0.756、0.690、0.704、0.701、0.737、0.884、0.991和0.961亿个rawreads。经过过滤后的测序数据分别为0.657、0.684、0.731、0.657、0.802、0.742、0.873、0.876、0.738、0.798、0.744、0.681、0.675、0.673、0.710、0.852、0.954和0.927亿个cleanreads。这些cleanreads的被拼接成217,836个转录本和161,095个unigene,分别合计2.571和2.406亿个核苷酸。transcript的平均长度为1,180bp,n50和n90分别为2,075bp和479bp;unigene的平均长度为1,494bp,n50和n90分别为2,203bp和714bp(表2)。
表2.小扁豆转录组测序组装结果.
(2)kasp标记的多态性分析
根据rna-seq结果,总共检测出130,073个snp。从中选取127个snp位点用于kasp标记的开发,并设计了127对kasp引物,最终有78个snp位点成功地转化为kasp标记,转化率为61.42%(表3)。利用78个kasp标记对94份小扁豆种质进行验证,其中76个标记是多态的,只有2个标记是单态的,分别占59.84%和1.57%。图1是引物wd_snp1基于sequencedetectionsystemsv2.4的检测结果。椭圆表示检测到的两个等位基因同为g,方框表示检测到的两个等位基因同为a,三角表示检测到的两个等位基因一个为g,另一个为a。表3结果显示,主要等位基因频率范围从0.5172到1.0000,平均值为0.8198;genotypenumber范围从1到3,平均值为2.7564;allelenumber范围从1到2,平均值为1.9744;genediversity范围从0到0.4994,平均值为0.2665;heterozygosity范围从0到0.9524,平均值为0.0846;pic范围从0到0.3747,平均值为0.2229。由此可得出,由snp到kasp标记的转化率较高,达到61.42%,因此与snp关联的性状可以很容易地转化为kasp标记,为将来用于小扁豆分子标记辅助育种提供支撑。
表3成功转化的78个kasp标记
(3)kasp标记的遗传多样性分析
pca分析
利用ntsys-pcv2.10e对全部参试的94份小扁豆种质进行基于kasp标记的pca分析,得到三维空间聚类图(图2)。前三维贡献率为79.35%。结果显示,中国资源(黑色椭圆)与国外资源(灰色椭圆)在空间上的分布是明显分离的,表明两者之间的亲缘关系较远,与地理来源显著相关。中国资源显著地聚集在一起,主要分布在聚类图的右侧,只有2份资源除外,这表明中国资源的遗传基础总体一致且较为狭窄,与国外资源显著不同;而国外资源呈现了宽广、均匀分布的空间特征,表明它们的遗传背景广泛且多样性分布较中国资源均匀。
upgma聚类分析
利用powermarkerv3.25计算获得遗传距离,结合megav6.0分析软件,对94份小扁豆种质(包括6份rna-seq种质和其余的88份验证种质)绘制基于kasp标记的upgma系统树图(图3),开放圆代表rna-seq的6份小扁豆资源;填充圆代表剩余的88份小扁豆种质;没有方框的圆代表中国小扁豆种质;被方框框出来的圆代表外国小扁豆资源。结果显示,94份小扁豆种质同样被分成了2大组群。第一组群内又分成了2个亚群,第一亚群全部是中国资源,第二亚群只有2份资源,分别来自日本和西亚的叙利亚。中国资源同样呈现两大特点与est-ssr标记一致,表明两种标记在区分中国资源时惊人的一致。第二组群同样被分成2个亚群,大多数为国外资源。第一亚群中只有2份资源,分别来自西亚的叙利亚和土耳其,第二亚群来源地较多,又被分成2个更小的分支,第一个分支中有5份资源,分别来自西亚的土耳其、北非的摩洛哥和北美的美国、加拿大,第二个分支也有5份资源,分别2份来自中国的陕西和新疆,2份来自西亚的土耳其和叙利亚,1份来自北美的加拿大。国外资源之间的遗传关系较远,表明其遗传基础更为宽泛。
群体结构分析
利用structurev2.3.4对94份小扁豆种质进行基于kasp标记的群体结构分析(图4)。结果显示,deltak(δk)值在k=2时数值最高,参试材料同样被分为2个亚群。其中深色表示第一亚群,有2份国外资源(a0000677来自日本,a0000279来自叙利亚);浅色表示第二亚群,大多数为国外资源,同样有2份中国资源(a0000170和a0000491),分别来自中国的陕西和新疆。此结果与pca分析和upgma聚类分析一致。
- 还没有人留言评论。精彩留言会获得点赞!