鲁拉西酮晶体及其制备方法和在注射给药系统中的应用与流程
本发明涉及医药
技术领域:
,具体涉及一种帕莫酸鲁拉西酮晶体、帕莫酸鲁拉西酮纳米晶体及其制备方法和应用。
背景技术:
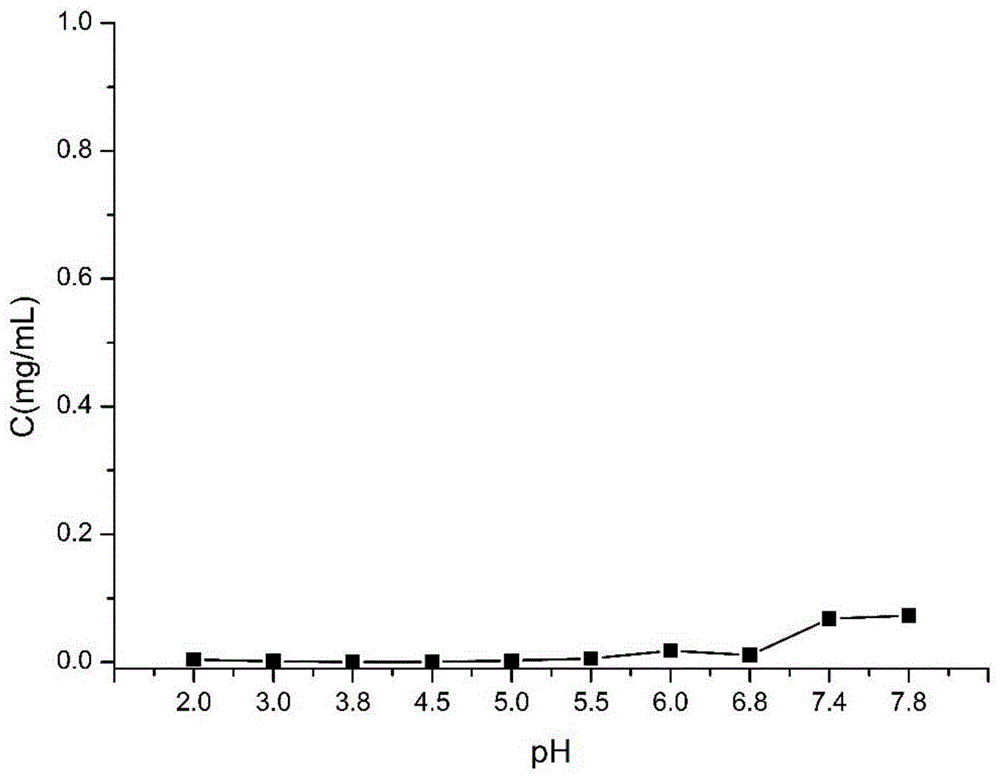
:盐酸鲁拉西酮(lurasidonehydrochloride)是由大日本住友制药发现的新型抗精神分裂症药物,2010年10月fda批准其口服片剂上市销售。盐酸鲁拉西酮与多巴胺d2受体和5-ht2a受体有着较高的亲和力,同时与5-ht7、5-ht1a和α2c肾上腺素受体亚型也有较高的亲和力。盐酸鲁拉西酮对精神分裂症患者的阳性症状和阴性症状均显示出良好的治疗效果。在有效治疗精神分裂症的同时避免了体重增加、锥体外系反应、qt间期延长等方面的不良反应。由于盐酸鲁拉西酮对5-ht7受体的拮抗作用,对记忆、情绪有着较好的调节作用;与α1去甲肾上腺素受体、组胺h1受体及胆碱能m1受体几乎没有亲和力,因此不需要剂量滴定。盐酸鲁拉西酮化学名称(3ar,4s,7r,7as)-2-[(1r,2r)-2-(4-(1,2-苯丙异噻唑-3-基)哌嗪-1-基甲基)环己基甲基]六氢-4,7-甲基异吲哚-1,3-二酮盐酸盐,为白色至类白色晶体粉末,略溶解于甲醇,微溶于乙醇,极微溶于水和丙酮,属生物药剂学分类里的bcsii药物,具有低溶解度、高渗透性的特性。盐酸鲁拉西酮溶解性受ph影响较大,在ph2.0-6.8范围内展现一定的ph依赖性,且在ph3.8处表现出最大溶解度,约0.78mg/ml。当ph﹥3.8时,随着ph的增大,溶解度越来越小,在ph6.8处溶解度仅0.007mg/ml。人体胃酸的ph约2.0,肠液ph约6.8,盐酸鲁拉西酮在胃肠道ph环境下较低的溶解度严重降低了其生物利用度,在人体中绝对生物利用度为9-19%。因此盐酸鲁拉西酮也是目前给药剂量(最大规格120mg)最大的精神分裂症治疗药物,并且临床使用时需与食物同服以提高吸收。目前提高鲁拉西酮溶解度的技术方案集中在制备新型的盐酸鲁拉西酮晶体或无定型。如中国发明专利申请201310066819.3公开了盐酸鲁拉西酮晶型及其制备方法和用途,但是该专利仅仅披露了所制备的盐酸鲁拉西酮晶体的特性参数及在储存和研磨等工序中无转晶现象,并未披露溶解度改善的数据。中国发明专利申请201310071135.2公开了盐酸鲁拉西酮a、b、c三种新晶型及其制备方法,其所制备的盐酸鲁拉西酮晶体在水和ph2.0酸性条件下的溶解度分别为3.4mg/ml和8.0mg/ml,溶解度差异约4.6mg/ml,差异很大,不符合理想注射剂原料释放速率应对ph不存在依赖性的特性。其次并未披露盐酸鲁拉西酮晶体在胃肠道ph范围内的溶解度情况,因而无法完整地体现其释放、吸收过程和饮食对它的影响以及对生物利用度的真正改善情况。技术实现要素:针对现有技术的不足,本发明提供了一种帕莫酸鲁拉西酮晶体、帕莫酸鲁拉西酮纳米晶体及其制备方法和应用。将鲁拉西酮制备成帕莫酸鲁拉西酮晶体,在ph2.0-7.8范围内溶解度基本无变化,在该ph范围内溶解度最大约0.07mg/ml;利用所制备的帕莫酸鲁拉西酮晶体制备注射液,能提高绝对生物利用度并具有长效缓释的效果。本发明提供了一种帕莫酸鲁拉西酮晶体,该帕莫酸鲁拉西酮晶体使用cu-kα辐射,以2θ表示的x射线粉末衍射光谱在9.327°、10.961°、15.296°、18.354°、18.801°、19.152°、21.185°、21.968°、23.096°、24.893°、25.989°、29.564°、33.771°处有衍射峰。优选地,所述帕莫酸鲁拉西酮晶体的溶解度在ph2-7.8之间为0.12-73μg/ml。优选地,所述帕莫酸鲁拉西酮晶体的溶解度在ph7-7.4之间为46-73μg/ml。本发明还提供了一种帕莫酸鲁拉西酮纳米晶体,该帕莫酸鲁拉西酮纳米晶体使用cu-kα辐射,以2θ表示的x射线粉末衍射光谱在9.696°、15.610°、17.274°、19.184°、19.461°、19.818°、20.407°、21.225°、22.099°、23.263°、24.672°、25.304°、26.305°、27.909°、29.356°、29.898°、32.171°、33.353°、35.015°、35.296°、36.160°、39.510°、40.426°、44.955°、54.213°、59.310°处有衍射峰。优选地,所述帕莫酸鲁拉西酮纳米晶体的粒径为250-500nm。本发明还提供了一种帕莫酸鲁拉西酮晶体的制备方法,该方法包括以下步骤:(1)将鲁拉西酮和帕莫酸溶解于四氢呋喃,得到第一混合溶液;(2)向所述第一混合溶液中加入乙酸乙酯,搅拌,加热反应后降至室温,得到第二混合溶液;(3)将所述第二混合溶液旋干后加入乙酸乙酯,二氯甲烷结晶,析出结晶后过滤,得到帕莫酸鲁拉西酮晶体。优选地,步骤(1)中鲁拉西酮和帕莫酸的摩尔比为1:1-1.2。优选地,步骤(2)中加热至55-60℃反应2-3h。本发明还提供了一种帕莫酸鲁拉西酮纳米晶体的制备方法,该方法包括以下步骤:(a)根据上述方法制备帕莫酸鲁拉西酮晶体;(b)将稳定剂溶解于纯化水中,再加入所述帕莫酸鲁拉西酮晶体,搅拌得到预混悬液;(c)将所述预混悬液加入高压均质机,在200-1000bar加压下循环,得到帕莫酸鲁拉西酮纳米晶体混悬液。优选地,所述方法还包括:(d)向所述帕莫酸鲁拉西酮纳米晶体混悬液中加入冻干保护剂,再放置在低温冰箱内冷冻;(e)将冷冻好的样品放置于冷冻干燥机,冷冻干燥,得到帕莫酸鲁拉西酮纳米晶体。优选地,所述稳定剂为泊洛沙姆188、聚维酮k12、十二烷基硫酸钠和十二烷基磺酸钠中的至少一种。优选地,所述泊洛沙姆188的质量浓度为0.1%(w/v)。优选地,步骤(c)中高压均质的压力为400-800bar。优选地,所述冻干保护剂为甘露醇,更优选为3-6重量%的甘露醇。优选地,步骤(d)中所述低温冰箱内的温度为-80±5℃。本发明还提供了一种药物组合物,该药物组合物含有上述帕莫酸鲁拉西酮晶体或者上述帕莫酸鲁拉西酮纳米晶体以及药学上可接受的辅料。优选地,所述药物组合物为帕莫酸鲁拉西酮注射液。本发明还提供了上述帕莫酸鲁拉西酮晶体或者上述帕莫酸鲁拉西酮纳米晶体在制备用于治疗精神分裂症的药物中的应用。与现有技术相比,本发明的上述技术方案具有以下优点:(1)本发明所述的帕莫酸鲁拉西酮晶体在ph2.0-7.8平衡溶解度为0.12-73μg/ml,溶解度绝对变化值小,总体呈平稳趋势,不受ph值的影响,不具有ph依赖性。(2)本发明所述的帕莫酸鲁拉西酮晶体在ph7.0-7.8(人体血液ph为7.1-7.4)平衡溶解度为46-73μg/ml,溶解度绝对变化值更小,受ph值影响小。(3)本发明所述的帕莫酸鲁拉西酮晶体在ph7.0-7.8溶解度小,最大也仅73μg/ml,具备了制备长效注射液的性质,为长效鲁拉西酮注射液创造了先决条件。(4)本发明所述的帕莫酸鲁拉西酮纳米晶体能提高帕莫酸鲁拉西酮的溶解度,增加药物的载药量;应用所制备帕莫酸鲁拉西酮纳米晶体制备的注射液具有缓释功能。附图说明图1所示为帕莫酸鲁拉西酮晶体平衡溶解度曲线图。图2所示为盐酸鲁拉西酮平衡溶解度曲线图。图3所示为帕莫酸鲁拉西酮晶体x-射线粉末衍射图。图4所示为帕莫酸鲁拉西酮纳米晶体x-射线粉末衍射图。图5所示为帕莫酸鲁拉西酮晶体差示扫描量热分析图。图6所示为帕莫酸鲁拉西酮纳米晶体差示扫描量热分析图。图7所示为帕莫酸鲁拉西酮纳米晶体粒径图。图8帕莫酸鲁拉西酮与帕莫酸鲁拉西酮纳米晶体体外释放曲线。具体实施方式以下对本发明的具体实施方式进行详细说明。应当理解的是,此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。本发明所述的帕莫酸鲁拉西酮晶体使用cu-kα辐射,以2θ表示的x射线粉末衍射光谱在9.327°、10.961°、15.296°、18.354°、18.801°、19.152°、21.185°、21.968°、23.096°、24.893°、25.989°、29.564°、33.771°处有衍射峰。在优选情况下,所述帕莫酸鲁拉西酮晶体的溶解度在ph2-7.8之间为0.12-73μg/ml,具体的例如可以为0.12μg/ml、0.25μg/ml、1.6μg/ml、2.3μg/ml、4μg/ml、5μg/ml、6μg/ml、8μg/ml、11μg/ml、18μg/ml、20μg/ml、25μg/ml、30μg/ml、35μg/ml、40μg/ml、46μg/ml、50μg/ml、55μg/ml、60μg/ml、68μg/ml、70μg/ml或73μg/ml。进一步优选地,所述帕莫酸鲁拉西酮晶体的溶解度在ph7-7.4之间为46-73μg/ml。在一种较优选的实施方式中,所述帕莫酸鲁拉西酮晶体为帕莫酸鲁拉西酮纳米晶体。所述帕莫酸鲁拉西酮纳米晶体使用cu-kα辐射,以2θ表示的x射线粉末衍射光谱在9.696°、15.610°、17.274°、19.184°、19.461°、19.818°、20.407°、21.225°、22.099°、23.263°、24.672°、25.304°、26.305°、27.909°、29.356°、29.898°、32.171°、33.353°、35.015°、35.296°、36.160°、39.510°、40.426°、44.955°、54.213°、59.310°处有衍射峰。进一步优选地,所述帕莫酸鲁拉西酮纳米晶体的粒径为250-500nm,具体的,例如可以为250nm、260nm、270nm、280nm、290nm、300nm、310nm、320nm、330nm、350nm、380nm、400nm、420nm、460nm、480nm或500nm。本发明所述的帕莫酸鲁拉西酮晶体的制备方法包括以下步骤:(1)将鲁拉西酮和帕莫酸溶解于四氢呋喃,得到第一混合溶液;(2)向所述第一混合溶液中加入乙酸乙酯,搅拌,加热反应后降至室温,得到第二混合溶液;(3)将所述第二混合溶液旋干后加入乙酸乙酯,二氯甲烷结晶,析出结晶后过滤,得到帕莫酸鲁拉西酮晶体。在步骤(1)中,鲁拉西酮和帕莫酸的摩尔比可以为1:1-1.2,最优选为1:1。在步骤(2)中,优选地,加热至55-60℃反应2-3h。本发明所述的帕莫酸鲁拉西酮纳米晶体的制备方法包括以下步骤:(a)根据上述方法制备帕莫酸鲁拉西酮晶体;(b)将稳定剂溶解于纯化水中,再加入所述帕莫酸鲁拉西酮晶体,搅拌得到预混悬液;(c)将所述预混悬液加入高压均质机,在200-1000bar加压下循环,得到帕莫酸鲁拉西酮纳米晶体混悬液。在较优选的实施方式中,所述方法还包括:(d)向所述帕莫酸鲁拉西酮纳米晶体混悬液中加入冻干保护剂,再放置在低温冰箱内冷冻;(e)将冷冻好的样品放置于冷冻干燥机,冷冻干燥,得到帕莫酸鲁拉西酮纳米晶体。在本发明中,所述稳定剂可以为本领域的常规选择。优选情况下,所述稳定剂为泊洛沙姆188、聚维酮k12、十二烷基硫酸钠和十二烷基磺酸钠中的至少一种。当选用泊洛沙姆188作为稳定剂时,优选地,所述泊洛沙姆188的质量浓度为0.1%(w/v)。在上述步骤(c)中,高压均质的压力可以为400-800bar,具体的例如可以为400bar、450bar、500bar、550bar、600bar、650bar、700bar、750bar或800bar。在上述步骤(d)中,所用冻干保护剂优选为甘露醇。更优选地,使用浓度为3-6重量%的甘露醇,最优选浓度为5重量%的甘露醇。在上述步骤(d)中,所述低温冰箱内的温度可以为-80±5℃,最优选为-80℃。本发明所述的药物组合物含有上述帕莫酸鲁拉西酮晶体或者上述帕莫酸鲁拉西酮纳米晶体以及药学上可接受的辅料。所述药学上可接受的辅料为本领域的常规选择。所述药物组合物的剂型没有特别的限定,可以为本领域熟知的各种剂型。在一种具体实施方式中,所述药物组合物为帕莫酸鲁拉西酮注射液。本发明所述的帕莫酸鲁拉西酮晶体或帕莫酸鲁拉西酮纳米晶体可用于治疗治疗精神分裂症。因而,所述帕莫酸鲁拉西酮晶体或所述帕莫酸鲁拉西酮纳米晶体可用于制备用于治疗精神分裂症的药物。以下实施例仅用于进一步说明本发明的内容,不应理解为对本发明的限制。在不背离本发明精神和实质的情况下,对本发明方法、步骤或条件所作的修改或替换,均属于本发明的范围。本发明实施例和对比例中所用原料均为市售产品。实施例1本实施例用于说明帕莫酸鲁拉西酮晶体的制备方法。将1g鲁拉西酮和788mg帕莫酸溶解于20ml四氢呋喃,再加入20ml乙酸乙酯,60℃反应2h后旋干。加入10ml乙酸乙酯和5ml二氯甲烷,待析出结晶后过滤得到产物帕莫酸鲁拉西酮a1。实施例2本实施例用于说明帕莫酸鲁拉西酮晶体的制备方法。将1g鲁拉西酮和788mg帕莫酸溶解于30ml四氢呋喃,再加入10ml乙酸乙酯,55℃反应3h后旋干。加入15ml乙酸乙酯和8ml二氯甲烷,待析出结晶后过滤得到产物帕莫酸鲁拉西酮a2。将产物在50℃真空干燥,并于玻璃瓶中室温、干燥、避光保存。实施例3本实施例用于说明帕莫酸鲁拉西酮纳米晶体的制备方法。称取0.1g泊洛沙姆188溶解于100ml纯化水中,再加入55mg帕莫酸鲁拉西酮a1,搅拌得到预混悬液;再将预混悬液加入高压均质机,在400bar加压下循环18次得到帕莫酸鲁拉西酮纳米晶体混悬液。实施例4本实施例用于制备帕莫酸鲁拉西酮注射液。配制帕莫酸鲁拉西酮注射液的处方如表1所示。表1帕莫酸鲁拉西酮晶体a110g聚山梨醇801g聚乙二醇40003g柠檬酸0.5g磷酸氢二钠0.5g氢氧化钠调ph值至6.8注射用水至100ml制备方法:取1g聚山梨酸酯80溶解于20ml注射用水中,将溶液通过0.2μm的无菌过滤器转移到已灭菌的不锈钢容器中;取帕莫酸鲁拉西酮晶体10g分散到40ml注射用水中,研磨4h后将悬浮液通过40μm无菌过滤器过滤到不锈钢容器中;取20ml注射用水,依次加入0.5g柠檬酸、0.5g磷酸氢二钠和3g聚乙二醇4000,混合均匀后通过0.2μm的无菌过滤器将溶液过滤到不锈钢容器中,混合均匀,并用无菌naoh溶液调ph至6.8后加注射用水至100ml,最后无菌灌装到无菌注射器中。实验例实验例1帕莫酸鲁拉西酮晶体的x射线粉末衍射分析测定方法:采用x-射线衍射仪cu-kα靶进行测定,电流40ma,电压40kv,扫描速度10°/min,步长0.02°,扫描范围3~60°,特征峰以2θ角度表示。测定结果:在9.327°、10.961°、15.296°、18.354°、18.801°、19.152°、21.185°、21.968°、23.096°、24.893°、25.989°、29.564°、33.771°处有特征峰。所得x-射线粉末衍射图如图3所示。x射线粉末衍射分析结果表明帕莫酸鲁拉西酮呈结晶状态。实验例2帕莫酸鲁拉西酮纳米晶体的x射线粉末衍射分析测定方法:采用x-射线衍射仪cu-kα靶进行测定,电流40ma,电压40kv,扫描速度10°/min,步长0.02°,扫描范围3~60°,特征峰以2θ角度表示。测定结果:在9.696°、15.610°、17.274°、19.184°、19.461°、19.818°、20.407°、21.225°、22.099°、23.263°、24.672°、25.304°、26.305°、27.909°、29.356°、29.898°、32.171°、33.353°、35.015°、35.296°、36.160°、39.510°、40.426°、44.955°、54.213°、59.310°处有特征峰。所得x-射线粉末衍射图如图4所示。实验例3帕莫酸鲁拉西酮晶体的差示扫描量热分析测定方法:取约2mg帕莫酸鲁拉西酮晶体放入铝锅里,扫描速率10℃/min,氮气流速为20ml/min,扫描温度范围25-400℃。测定结果:测得帕莫酸鲁拉西酮晶体在365℃处有放热峰。差示扫描量热分析结果表明帕莫酸鲁拉西酮晶体以晶体形式存在。差示扫描量热分析图谱如图5所示。实验例4帕莫酸鲁拉西酮纳米晶体的差示扫描量热分析测定方法:取约2mg帕莫酸鲁拉西酮晶体放入铝锅里,扫描速率10℃/min,氮气流速为20ml/min,扫描温度范围25-400℃。测定结果:测得帕莫酸鲁拉西酮纳米晶体在166℃处、365℃处有两个放热峰。差示扫描量热分析图谱如图6所示。实验例5帕莫酸鲁拉西酮晶体的红外光谱分析测定方法:分别取少量帕莫酸鲁拉西酮纳米晶体碾碎后,与kbr充分混合、研细,压片,置光谱仪扫描其红外吸收。红外光谱在400-4000cm-1波长范围内以4cm-1的解析度。测定结果:红外光谱在在3282cm-1附近出现的-nh键伸缩振动峰。2961cm-1-ch键伸缩振动峰。1250~1140cm-1处的吸收峰为c-c骨架振动峰。1680cm-1处的吸收峰为c=0(酰胺)伸缩振动峰。1600cm-1、1500cm-1处的吸收峰为芳环骨架的伸缩振动。1300~900cm-1处的吸收峰为c-s单键伸缩振动峰。860~800cm-1处的吸收峰为芳环的对二取代结构,770cm-1处的吸收峰为芳环的邻二取代结构。900~860cm-1,810~750cm-1,710~690cm-1处的吸收峰为芳环的间二取代结构。吸收峰1651cm-1处为双羟萘酸中游离羧酸c=o伸缩振动峰,1561cm-1处可观察到羧酸盐c=o伸缩振动峰。说明帕莫酸鲁拉西酮纳米晶体为鲁拉西酮新型晶体。实验例6帕莫酸鲁拉西酮晶体的核磁共振分析测定方法:取适量莫酸鲁拉西酮纳米晶体溶解于dmso-d6,再用300mhz核磁仪测定。测定结果:帕莫酸鲁拉西酮晶体在δ=0.9处甲基质子峰,δ=1.89处六元环亚甲基质子峰,δ=2.7~3.9处亚甲基不饱和碳碳双键的质子峰,δ=6.5~8处苯环的质子峰,δ=8.2处六元环酰胺的质子峰。δ=4.7处为双羟萘酸亚甲基的特征质子峰。δ=8.35处为伯胺盐的质子峰,未观察到游离羧基质子峰和游离伯氨基质子峰的化学位移。说明帕莫酸鲁拉西酮纳米晶体为鲁拉西酮新型晶体。实验例7帕莫酸鲁拉西酮晶体平衡溶解度的测定每次精密称取10mg帕莫酸鲁拉西酮晶体分别置于装有10mlph2.0、3.0、3.8、4.5、5.0、5.5、6.0、6.8、7.4、7.8磷酸盐缓冲液的玻璃瓶中,即得到1mg/ml帕莫酸鲁拉西酮溶液。将溶液置于37℃的水浴锅中搅拌,溶解平衡后24h取出样品,过0.22μm滤膜。取滤液稀释后测定其吸光度值,测定波长为235nm。测得的帕莫酸鲁拉西酮晶体在不同ph值溶液中的溶解度(μg/ml)如下表2所示。帕莫酸鲁拉西酮晶体平衡溶解度曲线图如图1所示。表2结果表明帕莫酸鲁拉西酮随着ph值的升高溶解度略有增加,最大溶解度约73μg/ml,总体呈平稳趋势;帕莫酸鲁拉西酮极难溶,并且在ph7.0-7.8平衡溶解度为46-73μg/ml,溶解度绝对变化值约27μg/ml,受ph值影响小。实验例8盐酸鲁拉西酮平衡溶解度的测定每次精密称取10mg盐酸鲁拉西酮分别置于装有10mlph2.0、3.0、3.8、4.5、5.0、5.5、6.0、6.8磷酸盐缓冲液的玻璃瓶中,即得到1mg/ml盐酸鲁拉西酮溶液。将溶液置于37℃的水浴锅中搅拌,溶解平衡后24h取出样品,过0.22μm滤膜。取滤液稀释后测定其吸光度值,测定波长为258nm。测得的盐酸鲁拉西酮在不同ph值溶液中的溶解度(mg/ml)如下表3所示。盐酸鲁拉西酮平衡溶解度曲线图如图2所示。表3ph2.03.03.84.55.05.56.06.8溶解度c(mg/ml)0.180.510.780.610.310.250.0120.007结果表明,在ph2.0-6.8的范围内,ph为3.8时,盐酸鲁拉西酮的溶解度最大;盐酸鲁拉西酮具有ph依赖性,在ph﹥3.8时,随着ph的增大,溶解度越来越小。实验例9帕莫酸鲁拉西酮纳米晶体的粒径及电位分析测定方法:将制备的帕莫酸纳米晶体混悬液用蒸馏水稀释至适宜浓度,采用马尔文粒度仪来测定平均粒径、多分散系数及zeta电位。纳米晶体冻干粉末复溶后采用上述相同的方法测定平均粒径、多分散系数及zeta电位。测得的帕莫酸鲁拉西酮纳米晶体的性质如表4所示。表4参数纳米晶体混悬液纳米晶体冻干粉末复溶后平均粒径(nm)298.2310.4pdi0.2570.231zeta电位(mv)-25.3-26.3制得的帕莫酸鲁拉西酮纳米晶体粒径在298.2nm左右。分布均匀且分布范围窄,pdi小于0.3。电位测定结果表明,纳米晶体表面带有负电荷,电位的绝对值大于25mv,表明体系电位比较稳定。纳米晶体冻干粉末复溶之后的粒径略有增加,其他参数没有显著变化。帕莫酸鲁拉西酮纳米晶体粒径图如图7所示。实验例10帕莫酸鲁拉西酮纳米晶体稳定性考察试验以粒径为评价指标,考察帕莫酸鲁拉西酮纳米混悬液在4℃条件下的物理稳定性,以初步确定帕莫酸鲁拉西酮长效注射液的贮存形式。同时考察帕莫酸鲁拉西酮纳米混悬液在37℃条件下的物理稳定性,以预测帕莫酸鲁拉西酮长效注射剂在体外释放的稳定性,结果如下表5所示。表5:帕莫酸鲁拉西酮在不同贮存条件下粒径和性状的变化情况实验结果表明帕莫酸鲁拉西酮纳米混悬液在4℃条件下贮存30天的物理稳定性良好,在随着放置时间的增加,粒径略有增加。37℃条件下贮存30天的帕莫酸鲁拉西酮纳米混悬液粒径与0时相比,平均粒径约增加了50nm物理稳定性良好,再分散性良好,可获得更平稳的药物释放度。实验例11不同形态的帕莫酸鲁拉西酮体外释放度本实验用透析袋法测定帕莫酸鲁拉西酮的体外释放度。精密称取501.1mg帕莫酸鲁拉西酮(标记为lpm)、粒径为286.4nm的帕莫酸鲁拉西酮冻干粉3.2g(约相当于原帕莫酸鲁拉西酮513.6mg,标记为ncps1),粒径为462.7nm的帕莫酸鲁拉西酮冻干粉5.3g(约相当于原帕莫酸鲁拉西酮501.8mg,标记为ncps2)。分别置于直径为22mm、长约10cm的透析袋中,除去袋内空气,两头用线绳扎紧,盘绕固定于溶出仪搅拌桨上,将搅拌桨置于距溶出杯底约1.5cm处,以用ph7.4的缓冲溶液900ml当溶出介质保持温度(37.0±0.5)℃,搅拌桨转速至50r/min,分别0.5h、1.0h、2.0h、4.0h、6.0h、8.0h、12.0h、24.0h、36.0h、48.0h、72h、144h、192h定时取样2ml(同时补加同体积的释放介质),用0.45μm微孔滤膜过滤,取续滤液进样,记录峰面积,计算出各时间帕莫酸鲁拉西酮的累计释放百分率。累计释药曲线以释放率为纵坐标,时间为橫坐标,详见图8。实验结果见下表6。释放度和累计释放百分率的计算公式如下:释放度(q)%=实际释放总量/药物总量×100%c为释放液的浓度,v为释放介质的体积,d为稀释倍数;w为药物的重量,f为药物中活性成分质量百分含量。表6:帕莫酸鲁拉西酮与帕莫酸鲁拉西酮纳米晶体的体外释放度结果表6的结果表明,粒径为286.4nm的帕莫酸鲁拉西酮纳米晶体(ncps1)的体外释放度大于帕莫酸鲁拉西酮原料药粉末(lpm)和粒径为462.7nm的帕莫酸鲁拉西酮纳米晶体(ncps2),说明将药物制备成纳米混悬液,由于粒径的减小,药物的溶解度和溶出速度都显著提高了。且粒径为286.4nm的帕莫酸鲁拉西酮纳米晶体冻干粉在每一时间段的释放量稳定,没有突释现象。说明帕莫酸鲁拉西酮纳米晶体中大部分为无定形结构,少部分多晶型结构,这比原料药中的大量多晶型结构有更高的溶解度及溶出速率,由于不同晶型的分子空间排列不同,造成了溶解度与溶出度的差异。而多晶型结构具有晶胞结构,具有较高的晶格能,将其变为分子态需要较高的能量,造成了其低溶解度及低溶出速率。但无定形的微观结构是分子或原子的无序集合,很容易溶解或溶出变成分子态,因此,帕莫酸鲁拉西酮纳米晶体提高了溶解度及体外释放速率。以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1