杂芳基雌激素受体调节剂及其用途的制作方法
本发明属于医药
技术领域:
,具体涉及杂芳基雌激素受体调节剂及其用途。
背景技术:
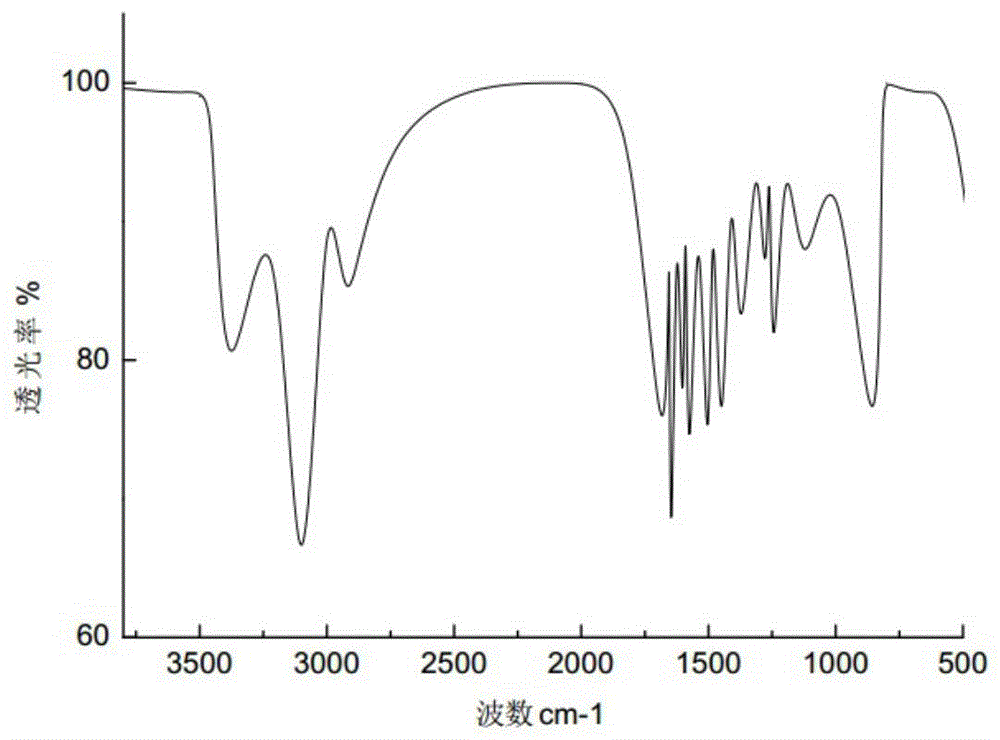
:雌激素受体(“ER”)是一种通过其与内源性雌激素相互作用而介导对多种生物效应的诱导的由配体活化的转录调节蛋白。内源性雌激素包括17β-雌二醇和雌酮。已发现ER具有两种亚型ER-α和ER-β。配体与亚型结合后,以不同的组织特异性发挥生理作用。雌激素和雌激素受体参与许多疾病或病症,如乳腺癌、肺癌、卵巢癌、结肠癌、前列腺癌、子宫内膜癌、子宫癌及其它疾病或病症。雌激素是人体中一类重要的激素化合物,当妇女进入绝经期后,体内雌激素水平下降,由此引发骨质疏松症、更年期综合征、老年痴呆症和心血管系统等疾病。针对绝经后雌激素水平下降,采取雌激素替代疗法或雌、孕激素联合使用的性激素替代疗法,能够显著降低绝经后骨质疏松性骨折和冠心病的发生率,以及克服雌激素致癌变的不良反应,但长期的HRT治疗仍可能增加乳腺癌的发生率,也并非在所有的情况下均能克服由雌激素所导致的子宫内膜癌的发生。选择性雌激素受体调节剂(selectiveestrogenreceptormodulators,SERMs)对骨骼和心血管系统具有雌激素样作用,而对子宫和乳腺表现出抗雌激素作用的药物。但是临床上使用的他莫西芬和雷洛昔芬能导致子宫内膜癌和热潮红等不良反应。因此,需要在转移性疾病和获得性耐药的情况下具有活性的新型雌激素受体-α靶向剂。现有技术如授权公告号为CN105732465B的中国发明专利公开了苯基吲哚类化合物及其制备方法和应用,并具体公开了苯基吲哚类化合物的通式如下:主要在制备治疗骨吸收性疾病药物中的应用,尤其是骨质疏松症。现有技术如授权公告号为CN105008382B的中国发明专利公开了用于抑制17β-羟类固醇脱氢酶(AKR1C3)的雌甾-1,3,5(10),16-四烯-3-羧酰胺,并具体公开了上述化合物的通式如下:上述化合物作为AKR1C3抑制剂,用于治疗和/或预防疾病的用途,尤其是月经障碍和子宫内膜异位症。技术实现要素:本发明的目的在于提供一种能作为雌激素受体降解剂、AKR1C3抑制剂、骨吸收抑制剂,用于治疗患者的雌激素受体相关疾病或病症的化合物以及包含该化合物和钙磷硅酸盐类微球载体的杂芳基雌激素受体调节剂,以及调节剂在治疗如乳腺癌、子宫内膜异位症及骨质疏松症上的用途。本发明的目的还在于提供一种能延长化合物保存期,增加微球产物负载量,增强调节剂稳定性和缓释性,在生物体液环境下提供过饱和量Ca2+与骨组织成核位点结合以促进骨组织的再生或修复的杂芳基雌激素受体调节剂的制备方法。本发明为实现上述目的所采取的技术方案为:本发明提供了通式(Ⅰ)的化合物:其中,R1选自氢、苯环、卤代苯、C1-C6烷基苯或C1-C6烷基苯基醚,X独立地为碳,其中所述碳能被R2取代,Y为碳或氮,其中所述碳能被R3取代,R2和R3,各自独立地为氢、卤素、C1-C6烷基、C1-C6烷氧基或C1-C6卤代烷基。本发明的化合物还包括通式(Ⅰ)化合物的立体异构体、互变异构体、药学上可接受的盐或上述物质的混合物;以及被通式(Ⅰ)包括的在下文中被指定为工作实施例的化合物,及其立体异构体、互变异构体、药学上可接受的盐或上述物质的混合物。本发明的化合物作为选择性雌激素受体调节剂(S雌激素受体M)的化合物,在某些实施例中,化合物可通过其作为雌激素受体降解剂的作用来解释,在基于细胞的测定中导致稳态ER-α水平的降低(即雌激素受体降解),且可用于治疗雌激素敏感性疾病或病症和/或已对抗激素疗法发展出抗性的疾病或病症;在另一些实施例中,化合物药效可通过其作为AKR1C3抑制剂的作用来解释;在另一些实施例中,化合物药效可作为骨吸收抑制剂的作用来解释。本发明还提供了杂芳基雌激素受体调节剂,该调节剂包括上述通式(Ⅰ)的化合物和微球载体;微球载体为钙磷硅酸盐类物质,其中二氧化硅、氧化钙和五氧化二磷的重量比为20:4:1,微球直径范围为300~1100nm。微球载体在肌体内是可生物降解的,利用微球作为化合物的载体,既能延长化合物保存期,又能引起调节剂在体内的缓释吸收。在某些具体实施方式中,调节剂中还包括药用载体、助流剂、稀释剂、赋形剂或药用治疗剂。上述药用治疗剂选自抗炎剂、免疫调节剂、细胞凋亡增强剂、组织蛋白酶K抑制剂、雄激素受体调节剂、破骨细胞质子ATP酶抑制剂、HMG-CoA还原酶抑制剂、整联蛋白受体拮抗剂、成骨细胞合成代谢剂例如PTH、降钙素、维生素D或合成的维生素D类似物、芳香酶抑制剂,及其药学上可接受的盐和混合物。本发明还提供了杂芳基雌激素受体调节剂的制备方法,包括,将上述通式(Ⅰ)的化合物分散于保护性胶体中形成分散体;将微球载体置于分散体中,于高压条件下混合,干燥,制得调节剂;上述高压条件的操作压力为4~10kg/cm2,接触时间为10~20min,然后在3~5s内恢复常压。在高压环境下,分散体分子之间联系紧密,得到优异的均质效果,通过瞬间将压力降为常压的方式,使得载体晶层在压力骤变中打开,层间间隙变大,而分散体也因此更深入地进入载体内部,不但有利于保护化合物活性,还能增强调节剂稳定性和缓释性,有利于调节剂后续深加工。在某些具体实施方式中,保护性胶体为藻酸盐形成的水凝胶,分散体中还包括药用载体、助流剂、稀释剂、赋形剂或药用治疗剂。在某些具体实施方式中,微球载体通过以下步骤获得:将十二胺溶于乙醇溶液中,然后添加正硅酸四乙酯、四水硝酸钙和磷酸三乙酯,水浴90~150min形成乳化液,然后陈化16~20h,洗涤所得沉淀,将沉淀冷冻干燥后,煅烧即得。优选地,乙醇溶液的体积浓度为70~85%,正硅酸四乙酯的添加量为十二胺重量的2~4倍,四水硝酸钙和磷酸三乙酯的添加量分别为十二胺重量的1~1.5倍和0.1~0.5倍。微球载体具有良好的生物相容性和生物活性,在体液中能形成弱碱性微环境起到抑菌作用,且玻璃微球中含有钙源、磷源,能与骨组织形成化学键合,预防或修复骨吸收性疾病引起的骨组织损伤。更进一步地,煅烧操作条件为:于300~500℃保温0.5~1h,然后升温至600~700℃保温1.5~2.5h。通过分段高温煅烧,使硅酸盐微核在递进式上升的温度中吸收能量配位层间发生畸变,硅离子与磷离子间局部排斥力减小,形成空间网格状结构,保温使得微核在该温度下持续性释放配位层间的应力,硅酸盐微核暴露出更多的可结合位点,以吸附更多Ca2+,并增加微球产物负载量,经过二次升温后,在更高的温度下,Ca2+向网格状晶体内部扩散,使得孔隙中堆积的Ca2+局部过饱和,在生物体液环境下过饱和Ca2+能脱离微球,与骨组织提供的成核位点结合,促进骨组织的再生或修复。本发明还提供了杂芳基雌激素受体调节剂的用途,其用于治疗患者的雌激素受体相关疾病或病症的方法,该方法包括向患有雌激素受体相关疾病或病症的患者给予治疗有效量的上述通式(Ⅰ)的化合物或上述制备方法制得的杂芳基雌激素受体调节剂。在某些具体实施方式中,雌激素受体相关疾病或病症是选自乳腺癌、肺癌、卵巢癌、子宫内膜癌、前列腺癌和子宫癌的癌症;在另一些具体实施方式中,雌激素受体相关疾病或病症是选自子宫内膜异位症、子宫出血紊乱、痛经、前列腺增生、多囊卵巢综合征或与炎症相关的疼痛;在另一些具体实施方式中,雌激素受体相关疾病或病症是选自骨丢失、骨折、牙周病、软骨退化、骨质疏松症的骨吸收性疾病。本发明的有益效果为:1)本发明提供了通式(Ⅰ)的化合物,作为选择性雌激素受体调节剂的化合物,可作为雌激素受体降解剂、AKR1C3抑制剂、骨吸收抑制剂,用于治疗患者的雌激素受体相关疾病或病症;2)本发明还提供了杂芳基雌激素受体调节剂,钙磷硅酸盐类物质作为化合物的微球载体,既能延长化合物保存期,又能引起调节剂在体内的缓释吸收;3)本发明微球载体制备中采用高压降压混合和分段高温煅烧,不但有利于保护化合物活性和增加微球产物负载量,还能增强调节剂稳定性和缓释性,并使得微球孔隙中堆积的Ca2+局部过饱和,并在生物体液环境下与骨组织提供的成核位点结合,促进骨组织的再生或修复;4)本发明的化合物及调节剂用于治疗患者的雌激素受体相关疾病或病症,如乳腺癌、肺癌、卵巢癌、子宫内膜癌、前列腺癌和子宫癌的癌症;子宫内膜异位症、子宫出血紊乱、痛经、前列腺增生、多囊卵巢综合征或与炎症相关的疼痛;骨丢失、骨折、牙周病、软骨退化、骨质疏松症的骨吸收性疾病。本发明采用了上述技术方案提供杂芳基雌激素受体调节剂及其用途,弥补了现有技术的不足,设计合理,操作方便。附图说明图1为实施例1所制化合物A的红外光谱示意图;图2为实施例2所制化合物B的红外光谱示意图;图3为化合物及调节剂对IL-6水平影响示意图。具体实施方式本发明提供了通式(Ⅰ)的化合物:其中,R1选自氢、苯环、卤代苯、C1-C6烷基苯或C1-C6烷基苯基醚,X独立地为碳,其中所述碳能被R2取代,Y为碳或氮,其中所述碳能被R3取代,R2和R3,各自独立地为氢、卤素、C1-C6烷基、C1-C6烷氧基或C1-C6卤代烷基。本发明的化合物还包括通式(Ⅰ)化合物的立体异构体、互变异构体、药学上可接受的盐或上述物质的混合物;以及被通式(Ⅰ)包括的在下文中被指定为工作实施例的化合物,及其立体异构体、互变异构体、药学上可接受的盐或上述物质的混合物。本发明的化合物作为选择性雌激素受体调节剂(S雌激素受体M)的化合物,在某些实施例中,化合物可通过其作为雌激素受体降解剂的作用来解释,在基于细胞的测定中导致稳态ER-α水平的降低(即雌激素受体降解),且可用于治疗雌激素敏感性疾病或病症和/或已对抗激素疗法发展出抗性的疾病或病症;在另一些实施例中,化合物药效可通过其作为AKR1C3抑制剂的作用来解释;在另一些实施例中,化合物药效可作为骨吸收抑制剂的作用来解释。在本发明的情形中优选的盐是本发明化合物的生理学可接受的盐,还涵盖这样的盐:其本身不适于药物应用,但是可用于例如本发明化合物的分离或纯化。本发明化合物的生理学可接受的盐包括无机酸、羧酸和磺酸的酸加成盐,包括但不限于盐酸、硫酸、磷酸、甲磺酸、乙磺酸、甲苯磺酸、苯磺酸、萘二磺酸、甲酸、乙酸、丙酸、乳酸、酒石酸、苹果酸、柠檬酸、富马酸、马来酸和苯甲酸。本发明化合物的生理学可接受的盐还包括常用碱的盐,包括但不限于碱金属盐(例如钠盐和钾盐)、碱土金属盐(例如钙盐和镁盐)。本申请化合物可通过适于待治疗的病状的任何途径来施用。合适的途径包括口服、胃肠外(包括皮下、肌内、静脉内、动脉内、皮内、鞘内和硬膜外)、经皮、直肠、经鼻、局部(包括含服和舌下)、阴道、腹膜内、肺内和鼻内。对于局部免疫抑制治疗,化合物可通过病灶内施用(包括灌注或在移植前使移植物与抑制剂接触)来施用。应该理解的是,优选的途径可随例如接受者的情况而变化。当所述化合物口服施用时,可将其与药用载体或赋形剂一起配制成丸剂、胶囊剂、片剂等。当所述化合物胃肠外施用时,可将其与药用胃肠外媒介物一起配制且配制成单位剂量注射形式。本发明还提供了杂芳基雌激素受体调节剂,该调节剂包括上述通式(Ⅰ)的化合物和微球载体;微球载体为钙磷硅酸盐类物质,其中二氧化硅、氧化钙和五氧化二磷的重量比为20:4:1,微球直径范围为300~1100nm。微球载体在肌体内是可生物降解的,利用微球作为化合物的载体,既能延长化合物保存期,又能引起调节剂在体内的缓释吸收。在某些具体实施方式中,微球载体与化合物的重量比为0.8~1.5:1。在某些具体实施方式中,调节剂中还包括药用载体、助流剂、稀释剂、赋形剂或药用治疗剂。合适的载体、稀释剂、添加剂和赋形剂是本领域技术人员已知的且包括但不限于例如碳水化合物、水溶性和/或溶胀性聚合物、乳糖、淀粉、蔗糖、葡萄糖、甲基纤维素、阿拉伯胶、明胶、油、水等物质,所使用的具体载体、稀释剂或赋形剂将取决于施用本发明化合物的手段和目的。上述药用治疗剂选自抗炎剂、免疫调节剂、细胞凋亡增强剂、组织蛋白酶K抑制剂、雄激素受体调节剂、破骨细胞质子ATP酶抑制剂、HMG-CoA还原酶抑制剂、整联蛋白受体拮抗剂、成骨细胞合成代谢剂例如PTH、降钙素、维生素D或合成的维生素D类似物、芳香酶抑制剂,及其药学上可接受的盐和混合物。在某些具体实施方式中,调节剂的给药形式包括但不限于:口服片剂,胶囊,栓剂,糖浆剂,颗粒剂,丸剂,散剂,乳剂,混悬剂,气雾剂等,这些剂型制备中可以添加包括溶剂、乳化剂和分散剂、粘合剂、着色剂、以及味道和/或气味矫正剂。本发明还提供了杂芳基雌激素受体调节剂的制备方法,包括,将上述通式(Ⅰ)的化合物分散于保护性胶体中形成分散体;将微球载体置于分散体中,于高压条件下混合,干燥,制得调节剂;上述高压条件的操作压力为4~10kg/cm2,接触时间为10~20min,然后在3~5s内恢复常压。在高压环境下,分散体分子之间联系紧密,得到优异的均质效果,通过瞬间将压力降为常压的方式,使得载体晶层在压力骤变中打开,层间间隙变大,而分散体也因此更深入地进入载体内部,不但有利于保护化合物活性,还能增强调节剂稳定性和缓释性,有利于调节剂后续深加工。在某些具体实施方式中,保护性胶体为藻酸盐形成的水凝胶,分散体中还包括药用载体、助流剂、稀释剂、赋形剂或药用治疗剂。在某些具体实施方式中,保护性胶体通过以下步骤获得:将藻酸盐加入到含有丙二醇的无菌水中,搅拌形成混合均匀的悬浮液,去除气泡即得;藻酸盐与丙二醇的重量比1:3~5,无菌水用量为藻酸盐的5~10倍量。藻酸盐、丙二醇均具有亲水性和吸湿保湿性,可结合水分形成水凝胶材料,使其他成分均匀地分散其中,利于微球负载化合物等成分使其不易被污染,延长货架期,同时作用于机体后,藻酸盐具有一定的渗出液吸收能力,能促进黏膜上创口愈合。在某些具体实施方式中,微球载体通过以下步骤获得:将十二胺溶于乙醇溶液中,然后添加正硅酸四乙酯、四水硝酸钙和磷酸三乙酯,水浴90~150min形成乳化液,然后陈化16~20h,洗涤所得沉淀,将沉淀冷冻干燥后,煅烧即得。优选地,乙醇溶液的体积浓度为70~85%,正硅酸四乙酯的添加量为十二胺重量的2~4倍,四水硝酸钙和磷酸三乙酯的添加量分别为十二胺重量的1~1.5倍和0.1~0.5倍。微球载体具有良好的生物相容性和生物活性,在体液中能形成弱碱性微环境起到抑菌作用,且玻璃微球中含有钙源、磷源,能与骨组织形成化学键合,预防或修复骨吸收性疾病引起的骨组织损伤。更进一步地,煅烧操作条件为:于300~500℃保温0.5~1h,然后升温至600~700℃保温1.5~2.5h。通过分段高温煅烧,使硅酸盐微核在递进式上升的温度中吸收能量配位层间发生畸变,硅离子与磷离子间局部排斥力减小,形成空间网格状结构,保温使得微核在该温度下持续性释放配位层间的应力,硅酸盐微核暴露出更多的可结合位点,以吸附更多Ca2+,并增加微球产物负载量,经过二次升温后,在更高的温度下,Ca2+向网格状晶体内部扩散,使得孔隙中堆积的Ca2+局部过饱和,在生物体液环境下过饱和Ca2+能脱离微球,与骨组织提供的成核位点结合,促进骨组织的再生或修复。本发明还提供了杂芳基雌激素受体调节剂的用途,其用于治疗患者的雌激素受体相关疾病或病症的方法,该方法包括向患有雌激素受体相关疾病或病症的患者给予治疗有效量的上述通式(Ⅰ)的化合物或上述制备方法制得的杂芳基雌激素受体调节剂。在某些具体实施方式中,雌激素受体相关疾病或病症是选自乳腺癌、肺癌、卵巢癌、子宫内膜癌、前列腺癌和子宫癌的癌症;在另一些具体实施方式中,雌激素受体相关疾病或病症是选自子宫内膜异位症、子宫出血紊乱、痛经、前列腺增生、多囊卵巢综合征或与炎症相关的疼痛;在另一些具体实施方式中,雌激素受体相关疾病或病症是选自骨丢失、骨折、牙周病、软骨退化、骨质疏松症的骨吸收性疾病。在下文中,为了帮助理解本发明,提供了以下实施例。然而,以下实施例仅用于使本发明更容易理解,而本发明的范围并不限制于在以下实例中。本发明还提供了以下方案来制备几个代表性实施例化合物:其中,R1选自氢、苯环、卤代苯、C1-C6烷基苯或C1-C6烷基苯基醚,X独立地为碳,其中所述碳能被R2取代,Y为碳或氮,其中所述碳能被R3取代,R2和R3,各自独立地为氢、卤素、C1-C6烷基、C1-C6烷氧基或C1-C6卤代烷基。实施例1:根据上述方案能制备得到具有通式(Ⅰ)的化合物A,其具体制备步骤如下:1)中间体1的制备:取4-羟基丙甲酮20g、无水碳酸钾100g溶于20倍量的丙酮中,然后向其中低级氯化苄22g,回流反应24h,减压蒸除溶剂丙酮,剩余物加水搅拌,抽滤、干燥,所得白色固体即为中间体1,m.p.101~102℃,ESI-MS:m/z241([M+H]+);2)中间体2的制备:取10g中间体1加入1,4-二氧六环和无水甲醇的混合溶剂中,缓慢滴加液溴7g,室温下反应至溶液透明澄清,加水搅拌1h,抽滤,干燥,所得白色固体即为中间体2,m.p.74~75℃,ESI-MS:m/z320([M+H]+);3)中间体3的制备:取6-硝基-1-茚满酮10g、氯化亚锡50g溶于20倍量的乙醇中,N2保护下,45℃反应24h,减压蒸除溶剂,向残留物中加入碳酸钠溶液,充分搅拌至不再有气泡产生,抽滤,滤饼加乙醇搅拌,抽滤,滤液旋蒸,所得白色固体即为中间体3,m.p.51~52℃,ESI-MS:m/z148([M+H]+);4)中间体4的制备:取8g中间体3、11g中间体2溶乙醇溶剂中,加入三乙胺7g,回流反应5h,自然冷却至室温,抽滤,所得乳白色固体即为中间体4,m.p.135~136℃,ESI-MS:m/z386([M+H]+);5)中间体5的制备:取12g中间体4、14g中间体3溶于乙二醇单乙醚中,回流反应6h,减压蒸除溶剂,剩余物加乙醇搅拌,抽滤,所得棕色固体即为中间体5,m.p.161~162℃,ESI-MS:m/z368([M+H]+);6)中间体6的制备:将5g中间体5溶于20倍量的二氯甲烷中,再添加5mL2,6-二叔丁基吡啶混合均匀后,向其中滴加3mL三氟甲磺酸酐,并将混合物在室温搅拌20小时,然后将混合物溶于碳酸氢钠饱和水溶液,搅拌混合物40min并分离各相,接着用二氯甲烷萃取两次,随后将合并的有机相用碳酸氢钠饱和溶液和氯化钠溶液洗涤,经硫酸钠干燥并浓缩,再通过与己烷搅拌进行萃取之后,即得中间体6,m.p.141~142℃,ESI-MS:m/z502([M+H]+);7)中间体7的制备:取8g中间体6和4g5-氟吡啶-3-硼酸添加在60mL甲苯和40mL乙醇中,再添加1.5g氯化锂、1g四三苯基膦钯和25mL碳酸钠饱和水溶液,并将混合物加热至100℃保持2.5h,然后将混合物用乙酸乙酯萃取三次,用碳酸氢钠饱和溶液和氯化钠溶液洗涤,并浓缩,在硅胶柱色谱法纯化(己烷/乙酸乙酯)后即得中间体7,m.p.102~103℃,ESI-MS:m/z447([M+H]+);8)中间体8的制备:取10g中间体7与3倍量丙酮搅拌溶解,再加入1:1的溴乙酸乙酯和碳酸钾混合溶液,与58℃下反应3h,然后将反应液倒入冰水中冷却后,得到棕色固体,最终通过柱层析提纯(洗脱剂为甲醇/二氯甲烷1:80),得到中间体8,m.p.89~90℃,ESI-MS:m/z443([M+H]+);9)中间体9的制备:取10g中间体8与3倍量乙醇搅拌溶解,再加入11mL水合肼,与78℃反应4h,再将反应液倒入冰水中冷却后,得到浅红色固体,最终通过柱层析提纯(洗脱剂为甲醇/二氯甲烷1:80),得到中间体9,m.p.118~120℃,ESI-MS:m/z429([M+H]+);10)化合物A的制备:取6g中间体9和N,N-二甲基甲酰胺二甲基缩醛1g溶解于50mL乙腈中,于60℃反应1h,然后加入7g芳香胺衍生物和6mL冰醋酸,于120℃回流24h,反应结束后,减压浓缩,剩余物用乙酸乙酯萃取,有机层用无水硫酸钠干燥,减压除去溶剂得到粗产物,最终通过柱层析提纯(洗脱剂为甲醇/二氯甲烷1:80),得到红褐色固体化合物A,结构式如下:m.p.79~80℃,ESI-MS:m/z438([M+H]+)。1HNMR(400MHz,CDCl3)δ7.65-7.59(m,4H),7.50-7.23(m,8H),7.26-6.99(m,2H),6.83-6.74(m,2H),5.96(d,J=2.9Hz,1H),5.54(d,J=3.4Hz,1H),4.86-4.76(m,1H),4.56-4.51(m,1H),4.45-4.38(m,1H),4.18-4.14(m,1H),3.98-3.85(m,2H),3.76-3.74(m,2H),3.69-3.54(m,1H),3.29-3.13(m,4H),3.01(q,J=7.3Hz,1H),2.86-2.74(m,1H),2.53-2.45(m,3H),1.39-1.22(m,1H),1.16(d,J=6.6Hz,3H),1.05-1.02(m,9H)。其红外光谱图见附图1。实施例2:根据上述方案能制备得到具有通式(Ⅰ)的红棕色固体化合物B,结构式如下:m.p.107~108℃,ESI-MS:m/z560([M+H]+),1HNMR(400MHz,CDCl3):δ7.48(d,J=7.6Hz,1H),7.22-7.16(m,1H),7.13-7.06(m,1H),6.92(d,J=10.5Hz,2H),5.96(s,1H),5.51(s,1H),5.88(d,J=5.0Hz,1H),4.53(t,J=5.7Hz,1H),4.41(t,J=6.0Hz,1H),4.27(dd,J=4.0,11.3Hz,1H),4.14-4.07(m,1H),3.84-3.80(m,1H,3.63(t,J=12.5Hz,2H),3.33-3.18(m,6H),2.93-2.78(m,2H),2.65-2.56(m,4H),1.75-1.67(m,2H),1.21-1.19(m,3H)。其红外光谱图见附图2。实施例3:杂芳基雌激素受体调节剂的制备方法,其具体步骤如下:1)将藻酸盐加入到6倍量含有丙二醇的无菌水中,搅拌形成混合均匀的悬浮液,去除气泡即得保护性胶体,藻酸盐与丙二醇的重量比为1:3.5;2)将化合物A加入保护性胶体中,搅拌均匀,形成分散体;3)将十二胺溶于体积浓度为75%的乙醇溶液中,然后添加正硅酸四乙酯、四水硝酸钙和磷酸三乙酯,水浴120min形成乳化液,然后陈化18h,洗涤所得沉淀,将沉淀冷冻干燥后,煅烧,即得微球载体,正硅酸四乙酯的添加量为十二胺重量的2.5倍,四水硝酸钙和磷酸三乙酯的添加量分别为十二胺重量的1.2倍和0.3倍,上述煅烧操作条件为:于450℃保温1h,然后升温至650℃保温2.5h;4)取化合物A重量1.2倍的微球载体置于分散体中,于操作压力为8kg/cm2的高压条件下接触20min,然后体系在5s内恢复常压,充分混合后,压片,干燥,制得片剂类调节剂。实施例4:本实施例与实施例3不同之处在于:步骤3)中煅烧操作条件为:升温至650℃保温3.5h,制得微球载体;其他步骤与实施例3中一致,制得片剂类调节剂。实施例5:本实施例与实施例3不同之处在于:步骤2)中加入的化合物为化合物B;其他步骤与实施例3中一致,制得片剂类调节剂。实施例6:本实施例与实施例3不同之处在于:步骤4)高压操作后的所得混合物中加入重量占比分别为20wt%的淀粉、5wt%的微晶纤维素和3wt%的硅氧烷流体,制得胶囊剂;其他步骤与实施例3中一致,制得胶囊类调节剂。实施例7:本实施例与实施例3不同之处在于:步骤4)高压操作后的所得混合物中加入羧甲基纤维素钠及糖浆混合以形成均匀糊状物,将苯甲酸溶液、矫味剂和着色剂用水稀释,并边搅拌边加入,然后加入足够量水以得到悬浮剂;其他步骤与实施例3中一致,制得悬浮剂类调节剂。试验例1:乳腺癌细胞增殖及雌激素受体降解试验试验样品:实施例1、2、3、5所制产物。1、乳腺癌细胞增殖测定(MCF-7):使MCF7扩增并维持于培养基中,将细胞调节至3000个细胞/mL的浓度,并且将细胞温育(37℃,5%CO2),次日,将10点连续稀释的样品以10-0.000005μM受试样品(17β-雌二醇用作对照)范围内的最终浓度添加至细胞。将附加细胞铺板到30孔中,以充当第1天(预处理)的比较。在样品暴露5天之后,将CellTiter-Glo试剂添加至细胞,并确定各个孔的相对发光单位(RLU)。另外将CellTiter-Glo添加至32μL不含细胞的培养基中,以获得背景值。让板在室温下温育10分钟,以使发光信号稳定,并用EnSpire记录发光信号。各个样品的细胞数的相对增加如下确定:(样品RLU-背景RLU/仅雌激素处理的细胞RLU-背景RLU)×100=抑制%。2、通过蛋白质印迹进行雌激素受体降解确定及ERαIC50确定:将MCF-7细胞以30万个细胞/mL(3mL/孔)铺板到6孔板的实验培养基中并在37℃,5%CO2下温育48小时。次日,在DMSO中制备化合物的10mM溶液,并将该溶液添加至细胞以实现10μM的最终浓度。对于IC50确定,将MCF-7细胞与3×或5×系列稀释的10mM化合物一起温育,化合物的最终浓度为10μM到基于化合物效能的设计浓度。DMSO对照包括在内,以允许确定受试化合物的相对功效。氟维司群用作ER-α降解的阳性对照,并且4-OH它莫西芬用作受体稳定性的对照。在将细胞与化合物一起温育18-24小时之后,制备细胞裂解物(2X细胞裂解缓冲液:100mMTris,pH8,300mMNaCl,2%NP40,1%脱氧胆酸钠,0.04%SDS,2mMEDTA)并且充分混合并在冰上温育。使用BCA试剂盒对蛋白质浓度进行定量。使用1×MES电泳缓冲液,在4-20%NuPAGENovex4-12%Bis-Tris蛋白质凝胶上分离蛋白质。然后将凝胶转移到硝酸纤维膜上。采用针对ESR1蛋白质的抗体(SantaCruz,sc-8005)探测印迹。GAPDH蛋白质用作内参。在AzureC600成像仪上使印迹成像,并且采用Azurespot软件定量蛋白质印迹的条带密度。采用GraphpadPrism计算IC50。结果如下表1:表1雌激素受体降解率及细胞增殖测定结果MCF-7IC50(nM)ERα降解率%实施例1AA实施例2AB实施例3AA实施例5AB对照组AA对于MCF-7IC50:A=单一IC50≤25nM,对于ERα降解率:A=ERα降解率>80%,B=ERα降解率<80%。由上表可知,本发明中化合物及调节剂在基于细胞的测定中导致稳态ER-α水平的降低,即雌激素受体降解,能作为雌激素受体降解剂,可用于治疗如乳腺癌等雌激素敏感性疾病或病症和/或已对抗激素疗法发展出抗性的疾病或病症。试验例2:化合物及调节剂的AKR1C3抑制作用测定试验样品:实施例1、2、3、5所制产物。试验方法:将样品溶于DMSO中,转移到黑色小体积384孔微量滴定板中,添加AKR1C3于测定缓冲液和蛋白酶抑制剂混合物溶液,并且将混合物孵育15min,然后通过添加NADPH和Coumberon于测定缓冲液中的溶液(3μl)来启动酶反应,并且将所得混合物在22℃孵育90min,使AKR1C3的浓度与酶制剂的相应活性匹配,并进行设置以使测定在线性范围内工作。典型的浓度处于1nM的区域。通过添加由抑制剂EM-1404组成的5μl终止液来终止反应。然后使用Pherastar在520nm(在380nm激发)测量Coumberol的荧光。荧光强度用作所形成的Coumberol的量的量度,因此用作AKR1C3的酶活性的量度。将数据归一化(没有抑制剂的酶反应=0%抑制;有所有其它测定组分但没有酶=100%抑制)。拟合计算IC50值。测定结果如下表2所示:表2化合物及调节剂的AKR1C3抑制结果AKR1C3酶抑制IC50(nM)实施例161实施例2119实施例383实施例5143对照组108由上表可知,化合物及调节剂对AKR1C3酶具有显著的抑制作用,可作为AKR1C3抑制剂使用。试验例3:化合物及调节剂对HOB细胞中IL-6和GM-CSF的作用测定试验样品:实施例1、2、3、4、5所制产物。试验方法:将人体破骨细胞HOB铺板在96孔皿内使其在常规HOB培养基中的密度为7×103个细胞/孔。次日,细胞用化合物或调节剂处理(0.2%DMSO)处理30分钟,随后加入IL-1β(1ng/mL)和TNF-α(10ng/mL),以未用化合物或调节剂处理的组别为空白组,培养持续24小时。利用市售ELISA试剂盒测定培养基中IL-6和GM-CSF水平。测定结果如下表3所示:表3化合物及调节剂对IL-6和GM-CSF的作用测定结果由上表可知,化合物及调节剂显示出对IL-6和GM-CSF的抑制作用,化合物及调节剂药效可作为骨吸收抑制剂使用;实施例4相较实施例3而言,其抑制作用表现较差,是由于微球载体煅烧方式的不同导致微球产物的负载量不同,同时微球与骨组织提供的成核位点结合量不同,故而表现出不同的效果。图3为化合物及调节剂对IL-6水平影响示意图,由图中可知,实施例1、3、4所制的化合物及调节剂在24小时培养时间内,较空白组能有效的抑制IL-6水平上升,实施例1和3在24小时内均能有效达到抑制效果,实施例4在前18小时抑制效果也较为突出,但后6小时抑制效果明显下降,是由于微球载体煅烧方式的不同导致实施例4微球产物的负载量较小,有效物质的量较小,使得药物效果相较其他而言较差。上述实施例中的常规技术为本领域技术人员所知晓的现有技术,故在此不再详细赘述。以上实施方式仅用于说明本发明,而并非对本发明的限制,本领域的普通技术人员,在不脱离本发明的精神和范围的情况下,还可以做出各种变化和变型。因此,所有等同的技术方案也属于本发明的范畴,本发明的专利保护范围应由权利要求限定。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1