一种肉桂酸衍生物及其制备方法和应用与流程
本发明涉及药物化学领域,具体涉及一种肉桂酸衍生物及其制备方法和应用。
背景技术:
在过去几十年里,化学农药的使用为农业的发展起到了重要的作用,常规化学农药存在残留、抗药性及环境安全性等问题。随着时间的推移,常规化学农药存在的问题日益突显出来,1962年美国海洋生物学家carson女士的著作《寂静的春天》出版后,披露了大量有毒农药品种如甲胺磷、对硫磷、磷胺等在使用过程中杀死有益生物,污染土壤,破坏生态等严重问题,激起人们对使用化学农药后产生的环境问题高度重视。
肉桂酸类化合物属于苯丙素类的天然化合物,普遍地存在于肉桂、蔬菜和水果蜂胶中,属于天然的抗氧化剂,且具有良好的抗氧化、抗菌、抗肿瘤、杀虫活性等优点。因此,它被广泛地应用于医药上、农业上。另外,硝基苯乙烯类化合物结构中含有
n-溴代丁二酰亚胺在农业中常被用来合成一种有效的噻菌灵来防治作物的真菌病害。亚磷酸二烷基酯是一种重要有机合成中间体,具有很好的抗磨性和抗氧化性,在有机磷农药制备占用重要地位。
基于此,结合肉桂酸类化合物和不同的取代基化合物的优点开发出一种低毒、低残留、高活性的环境友好型的肉桂酸衍生物来替代过去的高毒、高残留农药具有重大意义。
技术实现要素:
为解决上述问题,本发明提供了一种肉桂酸衍生物,所述衍生物为式i所示化合物或式i所示化合物的对应异构体、非对映异构体、外消旋体、结晶水合物或溶剂合物,
其中,r1表示任意取代的烷氧基、氢基、卤素或任意取代的磺酰基;
r2表示任意取代的酯基、1-4个碳原子的烷基或氢;
r3表示卤素或硝基;
x表示卤素;
优选地,所述r1为-och3、-cl、-f、-br、-h、甲基磺酰基;
所述r2为
所述r3为-br或-no2;
所述x为-br、-cl、-f。
更优选地,所述肉桂酸衍生物为如下所示化合物:
本发明还提供了肉桂酸衍生物的制备方法,
当
s11使化合物a与化合物b接触,获得式c所示化合物,
s12使化合物c与化合物d接触,以便获得e所示化合物;
当
s21使化合物a与化合物f接触,获得式g所示化合物,
s22使化合物g与卤素单质接触,以便获得h所示化合物;
优选地,
当
s111化合物a、化合物b和第一催化剂溶解于第一溶剂中,加热回流4-10小时,得到化合物c,
s121化合物c、化合物d和第二催化剂溶解于在第二溶剂中,加热回流7-8小时,得到化合物e;
当
s211化合物f、化合物g和第三催化剂溶解于第三溶剂中,加热回流5-10小时,得到化合物h,
s221化合物h、卤素单质和第四催化剂溶解于第四溶剂中,得到化合物i。
所述第一催化剂为吡啶,
所述第一溶剂为n,n-二甲基甲酰胺、三氯甲烷、丙酮、二氯甲烷,
所述第二催化剂为四甲基胍,
所述第二溶剂为冰醋酸、二氯甲烷、三氯甲烷、丙酮、乙醇、甲醇,
所述第三催化剂为三乙胺、乙酸钠、乙酸铵,
所述第三溶剂为二氯甲烷、三氯甲烷,丙酮、乙酸,
所述第四催化剂为乙酸钠,
所述第四溶剂为冰醋酸。
所述化合物a、化合物b、第一催化剂、第一溶剂的配比为(8-12)mmol:(25-35)mmol:(8-12)mmol:(15-30)ml,
所述化合物c、化合物d、第二催化剂和第二溶剂的配比为(8-12)mmol:(15-30)mmol:(0.5-2)mmol:(15-30)ml,
所述化合物f、化合物g、第三催化剂和第三溶剂的配比为(8-12)mmol:(25-35)mmol:10mmol:(15-30)ml,
所述化合物h、卤素单质、第四催化剂和第四溶剂的配比为(8-12)mmol:(8-15)mmol:12mmol:(15-30)ml。
此外,本发明还提供一种农药,包括上述定义的化合物。
本发明提供了一种杀菌方法,为农作物施加上述的化合物或农药,所述农作物为水稻、小麦、果树、蔬菜。
本发明开发出一种低毒、低残留、高活性的环境友好型肉桂酸衍生物,所述肉桂酸衍生物农药,可替代传统的高毒、高残留农药。
附图说明
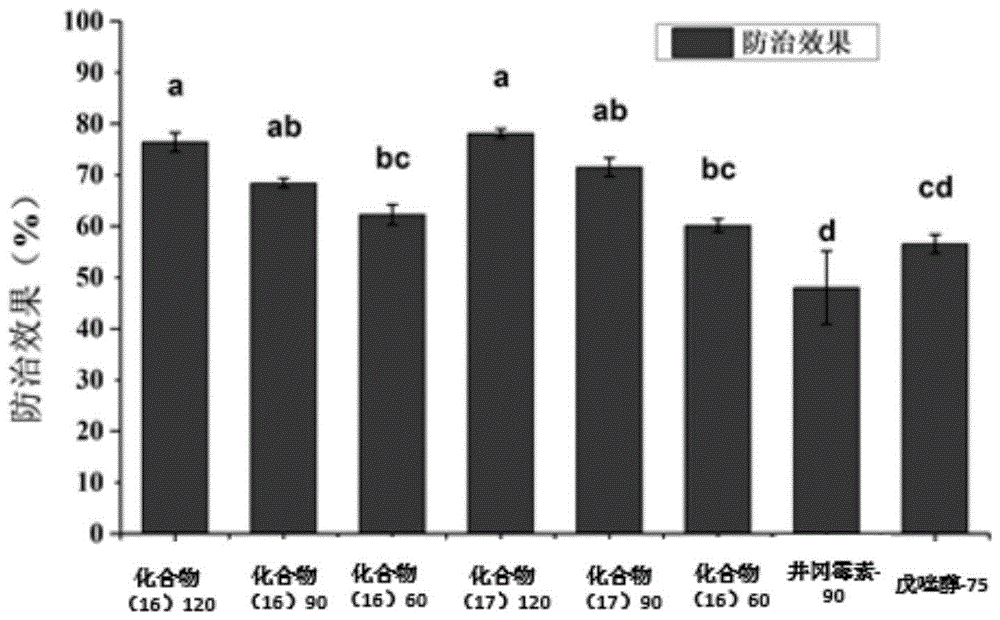
图1化合物(16)和(17)对水稻纹枯病(r.solani)的防治效果图
具体实施例
下面通过实例来具体地说明本发明中肉桂酸衍生物的制备方法,这些实施例仅对本发明进行说明,而不是对本发明进行限制。
方法一:
以取代苯甲醛、丙二酸为原料,吡啶作为催化剂,n,n-二甲基甲酰胺、三氯甲烷、丙酮、二氯甲烷等有机试剂作为溶剂条件下,取代苯甲醛与丙二酸发生反应生成中间体c;中间体c与一分子的nbs发生汉斯狄克反应生成烯溴,烯溴再与一分子的nbs反应,最终制得化合物e。
向100ml单颈圆底烧瓶中,依次加入不同取代的苯甲醛(8-12mmol),丙二酸25-35mmol,吡啶8-12mmol,15-30ml的n,n-二甲基甲酰胺或三氯甲烷或丙酮或二氯甲烷。加热150℃回流6-7h;向反应体系加入20ml水,随后用5%稀hcl调至ph=1,大量固体析出,抽滤,得化合物c。
向100ml单颈圆底烧瓶中,依次加入化合物c(8-12mmol),nbs(15-30mmol),四甲基胍(0.5-2mmol),15-30ml冰醋酸、二氯甲烷、三氯甲烷、丙酮、乙醇、甲醇等溶剂,加热回流7-8h;加入饱和的nahco3溶液,调至ph=1,拌样,柱层析,展开剂为石油醚:乙酸乙酯=1:50或1:30或1:40或1:60,得到目标化合物e。
r1表示任意取代的烷氧基、氢、卤素或任意取代的磺酰基;
r2表示任意取代的酯基、1-4个碳原子的烷基或氢;
r3表示卤素或硝基;
x表示卤素。
方法二:
以取代苯甲醛为原料,三乙胺,乙酸钠或乙酸铵作为催化剂,二氯甲烷、三氯甲烷、丙酮或乙酸为溶剂条件下,取代苯甲醛与硝基甲烷发生反应生成化合物h;化合物h与卤素单质发生α取代,最终制得化合物i。
向100ml单颈圆底烧瓶中,加入取代苯甲醛(8-12mmol),硝基甲烷(25-35mmol),催化剂用三乙胺、乙酸钠或乙酸铵(8-12mmol),溶剂用二氯甲烷、乙酸、三氯甲烷或丙酮(15-30ml)。150℃加热回流5-10h,tlc检测反应至结束。向反应体系加入约50ml水,二氯甲烷萃取,合并有机相,无水硫酸钠干燥;抽滤,拌样,乙酸乙酯和石油醚为洗脱剂,洗脱剂可以选用ea:pe=1:40,1:45,1:50,1:60,柱层析分离纯化得化合物h。
向100ml单颈圆底烧瓶中,依次加入化合物h(8-12mmol),卤素单质(8-15mmol),乙酸钠(8-20mmol),(15-30)ml冰醋酸或二氯甲烷或三氯甲烷或丙酮或乙醇或甲醇,tlc检测试验至反应结束;加入约30ml的饱和的硫代硫酸钠,除去过量的液溴;二氯甲烷萃取,合并有机相,无水硫酸钠干燥;抽滤,拌样,柱层析,洗脱剂为石油醚:乙酸乙酯=1:50,最终得到目标化合物i。
r1表示任意取代的烷氧基、氢、卤素或任意取代的磺酰基;
r2表示任意取代的酯基、1-4个碳原子的烷基或氢;
r3表示卤素或硝基;
x表示卤素。
实施例1
向100ml单颈圆底烧瓶中,依次加入烷氧基对位取代的苯甲醛8mmol,丙二酸25mmol,吡啶8mmol,15ml的n,n-二甲基甲酰胺。加热150℃回流6-7h;向反应体系加入20ml水,随后用5%稀hcl调至ph=1,在调至的过程中,大量固体析出,抽滤,得化合物c1。
向100ml单颈圆底烧瓶中,依次加入8mmol化合物c1,nbs15mmol,四甲基胍0.5mmol,15ml冰醋酸,加热回流7-8h;加入饱和的nahco3溶液,调至ph=1,拌样,柱层析,展开剂为石油醚:乙酸乙酯=1:50,得到化合物(1)。
所得化合物(1)为淡黄色固体,收率为88%。mp.51~53℃;
元素分析:实测值c%37.29h%3.13,计算值c%37.53h%3.44;
1hnmr(400mhz,dmso-d6,tms)δ(ppm):7.38(d,j=7.8hz,2h),6.92(d,j=7.9hz,2h),6.50(d,j=4.4hz,1h),6.06(d,j=4.4hz,1h),3.75(s,3h),2.15(s,3h);
13cnmr(100mhz,dmso-d6,tms)δ(ppm):173.86,164.50,133.74,132.99,118.76,82.74,60.38,53.47,25.93;
ei-ms(70ev)m/z(%):350[m]+,198(38.30),154(36.88),212(12.71),213(100.0),111(17.9);
实施例2
向100ml单颈圆底烧瓶中,依次加入氯对位取代的苯甲醛10mmol,丙二酸20mmol,吡啶10mmol,20ml的n,n-二甲基甲酰胺。加热150℃回流6-7h;向反应体系加入20ml水,随后用5%稀hcl调至ph=1,在调至的过程中,大量固体析出,抽滤,得化合物c2。
向100ml单颈圆底烧瓶中,依次加入10mmol化合物c2,nbs20mmol,四甲基胍1mmol,20ml冰醋酸,加热回流7-8h;加入饱和的nahco3溶液,调至ph=1,拌样,柱层析,展开剂为石油醚:乙酸乙酯=1:50,得到化合物(2)。
所得化合物(2)为白色固体,收率为90%。mp.54~555℃;
元素分析:实测值c%34.09h%2.83,计算值c%33.70h%2.55;
1hnmr(400mhz,dmso-d6,tms)δ(ppm):7.57–7.26(m,3h),6.52(d,j=4.4hz,1h),6.10(d,j=4.4hz,1h),2.15(s,3h);
13cnmr(100mhz,dmso-d6,tms)δ(ppm):173.90,140.07,138.57,134.17,133.36,82.14,52.63,25.87;
ei-ms(70ev)m/z(%):356[m]+,143(49.88),183(29.40),195(9.89),141(100.0),275(8.85);
实施例3
向100ml单颈圆底烧瓶中,依次加入氟对位取代的苯甲醛10mmol,丙二酸30mmol,吡啶10mmol,20ml的三氯甲烷。加热150℃回流6-7h;向反应体系加入20ml水,随后用5%稀hcl调至ph=1,在调至的过程中,大量固体析出,抽滤,得化合物c3。
向100ml单颈圆底烧瓶中,依次加入10mmol化合物c3,nbs20mmol,四甲基胍1mmol,20ml冰醋酸,加热回流7-8h;加入饱和的nahco3溶液,调至ph=1,拌样,柱层析,展开剂为石油醚:乙酸乙酯=1:50,得到化合物(3)。
所得化合物(3)为淡黄色液体,收率为87%。n=1.5483;
元素分析:实测值c%35.62h%2.83,计算值c%35.33h%2.67;
1hnmr(400mhz,dmso-d6,tms)δ(ppm):7.47(d,j=5.2hz,2h),7.17(t,j=8.7hz,2h),6.51(d,j=4.4hz,1h),6.11(d,j=4.4hz,1h),2.14(s,3h);
实施例4
向100ml单颈圆底烧瓶中,依次加入氟邻位取代的苯甲醛10mmol,丙二酸30mmol,吡啶10mmol,20ml的二氯甲烷。加热150℃回流6-7h;向反应体系加入20ml水,随后用5%稀hcl调至ph=1,在调至的过程中,大量固体析出,抽滤,得化合物c4。
向100ml单颈圆底烧瓶中,依次加入10mmol化合物c4,nbs20mmol,四甲基胍1mmol,20ml冰醋酸,加热回流7-8h;加入饱和的nahco3溶液,调至ph=1,拌样,柱层析,展开剂为石油醚:乙酸乙酯=1:50,得到化合物(4)。
所得化合物(4)为淡黄色液体,收率为91%。n=1.6402;
元素分析:实测值c%35.59h%2.83,计算值c%35.33h%2.67;
1hnmr(400mhz,dmso-d6,tms)δ(ppm):7.64–6.98(m,4h),6.52(d,j=6.4hz,1h),6.27(d,j=6.4hz,1h),2.11(s,3h);
实施例5
向100ml单颈圆底烧瓶中,依次加入溴对位取代的苯甲醛10mmol,丙二酸30mmol,吡啶10mmol,20ml的n,n-二甲基甲酰胺。加热150℃回流6-7h;向反应体系加入20ml水,随后用5%稀hcl调至ph=1,在调至的过程中,大量固体析出,抽滤,得化合物c5。
向100ml单颈圆底烧瓶中,依次加入10mmol化合物c5,nbs20mmol,四甲基胍1mmol,20ml冰醋酸,加热回流7-8h;加入饱和的nahco3溶液,调至ph=1,拌样,柱层析,展开剂为石油醚:乙酸乙酯=1:50,得到化合物(5)。
所得化合物(5)为淡黄色固体,收率为88%。m.p.68-70℃;
元素分析:实测值c%35.59h%2.83,计算值c%29.96h%2.26;
1hnmr(400mhz,dmso-d6,tms)δ(ppm):7.54(t,j=8.3hz,2h),7.38(d,j=8.1hz,2h),6.51(s,j=4.4hz,1h),6.08(s,j=4.4hz,1h),2.15(s,3h);
13cnmr(100mhz,dmso-d6,tms)δ(ppm):173.89,140.48,136.28,134.45,127.26,82.21,52.51,25.88;
ei-ms(70ev)m/z(%):398[m]+,183(46.55),204(27.38),259(23.68),102(100.0),227(26.48);
实施例6
向100ml单颈圆底烧瓶中,依次加入氯对位和邻位取代的苯甲醛10mmol,丙二酸30mmol,吡啶10mmol,20ml的n,n-二甲基甲酰胺。加热150℃回流6-7h;向反应体系加入20ml水,随后用5%稀hcl调至ph=1,在调至的过程中,大量固体析出,抽滤,得化合物c6。
向100ml单颈圆底烧瓶中,依次加入10mmol化合物c6,nbs20mmol,四甲基胍1mmol,20ml冰醋酸,加热回流7-8h;加入饱和的nahco3溶液,调至ph=1,拌样,柱层析,展开剂为石油醚:乙酸乙酯=1:50,得到化合物(6)。
所得化合物(6)为淡黄色固体,收率为85%。m.p.70-72℃;
元素分析:实测值c%31.95h%2.56,计算值c%30.73h%2.06;
1hnmr(400mhz,dmso-d6,tms)δ(ppm):7.74–7.35(m,3h),6.52(d,j=4.8hz,1h),6.35(d,j=4.8hz,1h),2.13(s,3h);
ei-ms(70ev)m/z(%):388[m]+,185(90.94),229(40.22),174(100.0),275(13.79);
实施例7
向100ml单颈圆底烧瓶中,依次加入烷氧基间位取代的苯甲醛10mmol,丙二酸30mmol,吡啶10mmol,20ml的n,n-二甲基甲酰胺。加热150℃回流6-7h;向反应体系加入20ml水,随后用5%稀hcl调至ph=1,在调至的过程中,大量固体析出,抽滤,得化合物c7。
向100ml单颈圆底烧瓶中,依次加入10mmol化合物c7,nbs20mmol,四甲基胍1mmol,20ml冰醋酸,加热回流7-8h;加入饱和的nahco3溶液,调至ph=1,拌样,柱层析,展开剂为石油醚:乙酸乙酯=1:50,得到化合物(7)。
所得化合物(7)为淡黄色固体,收率为90%。m.p.52-54℃;
元素分析:实测值c%37.23h%3.19,计算值c%37.53h%3.44;
1hnmr(400mhz,dmso-d6,tms)δ(ppm):7.25(d,j=7.8hz,1h),7.14(d,j=7.8hz,1h),6.92(d,j=7.9hz,2h),6.50(d,j=4.4hz,1h),6.06(d,j=4.4hz,1h),3.83(s,3h),2.15(s,3h);
13cnmr(100mhz,dmso-d6,tms)δ(ppm):172.86,164.50,133.74,132.99,119.76,82.74,60.20,52.47(s),25.82;
ei-ms(70ev)m/z(%):350[m]+,196(14.24),212(20.08),290(100.0),367(20.66);
实施例8
向100ml单颈圆底烧瓶中,依次加入氯间位取代的苯甲醛10mmol,丙二酸30mmol,吡啶10mmol,20ml的n,n-二甲基甲酰胺。加热150℃回流6-7h;向反应体系加入20ml水,随后用5%稀hcl调至ph=1,在调至的过程中,大量固体析出,抽滤,得化合物c8。
向100ml单颈圆底烧瓶中,依次加入10mmol化合物c8,nbs20mmol,四甲基胍1mmol,20ml冰醋酸,加热回流7-8h;加入饱和的nahco3溶液,调至ph=1,拌样,柱层析,展开剂为石油醚:乙酸乙酯=1:50,得到化合物(8)。
所得化合物(8)为淡黄色固体,收率为95%。m.p.30-31℃;
元素分析:实测值c%33.50h%2.83,计算值c%33.70h%2.55;
1hnmr(400mhz,dmso-d6,tms)δ(ppm):7.45(dd,j=55.9,22.0hz,5h),6.57(d,j=4.4hz,1h),6.15(d,j=4.4hz,1h),2.20(s,3h);
实施例9
向100ml单颈圆底烧瓶中,依次加入氯邻位取代的苯甲醛10mmol,丙二酸30mmol,吡啶10mmol,20ml的n,n-二甲基甲酰胺。加热150℃回流6-7h;向反应体系加入20ml水,随后用5%稀hcl调至ph=1,在调至的过程中,大量固体析出,抽滤,得化合物c9。
向100ml单颈圆底烧瓶中,依次加入10mmol化合物c9,nbs20mmol,四甲基胍1mmol,20ml冰醋酸,加热回流7-8h;加入饱和的nahco3溶液,调至ph=1,拌样,柱层析,展开剂为石油醚:乙酸乙酯=1:50,得到化合物(9)。
所得化合物(9)为淡黄色固体,收率为91%。m.p.75-77℃;
元素分析:实测值c%33.59h%2.83,计算值c%33.70h%2.55;
1hnmr(400mhz,dmso-d6,tms)δ(ppm):7.61(s,1h),7.51(s,1h),7.43(s,1h),7.28(s,1h),6.46(d,j=4.4hz,1h),6.38(d,j=4.4hz,1h),2.15(s,3h);
ei-ms(70ev)m/z(%):356[m]+,155(11.86),181(16.82),184(100.0),318(18.52);
实施例10
向100ml单颈圆底烧瓶中,依次加入溴邻位取代的苯甲醛10mmol,丙二酸30mmol,吡啶10mmol,20ml的n,n-二甲基甲酰胺。加热150℃回流6-7h;向反应体系加入20ml水,随后用5%稀hcl调至ph=1,在调至的过程中,大量固体析出,抽滤,得化合物c10。
向100ml单颈圆底烧瓶中,依次加入10mmol化合物c10,nbs20mmol,四甲基胍1mmol,20ml冰醋酸,加热回流7-8h;加入饱和的nahco3溶液,调至ph=1,拌样,柱层析,展开剂为石油醚:乙酸乙酯=1:50,得到化合物(10)。
所得化合物(10)为白色固体,收率为95%。m.p.53-55℃;
元素分析:实测值c%30.19h%2.43,计算值c%29.96h%2.26;
1hnmr(400mhz,dmso-d6,tms)δ(ppm):7.59–7.34(m,4h),6.50(d,j=5.3hz,1h),6.43(d,j=5.3hz,1h),2.16(s,3h);
ei-ms(70ev)m/z(%):398[m]+,138(17.00),184(20.43),140(100.0),320(13.78);
实施例11
向100ml单颈圆底烧瓶中,依次加入氯对位和间位取代的苯甲醛8mmol,丙二酸35mmol,吡啶12mmol,30ml的n,n-二甲基甲酰胺。加热150℃回流6-7h;向反应体系加入20ml水,随后用5%稀hcl调至ph=1,在调至的过程中,大量固体析出,抽滤,得化合物c11。
向100ml单颈圆底烧瓶中,依次加入12mmol化合物c11,nbs30mmol,四甲基胍2mmol,30ml冰醋酸,加热回流7-8h;加入饱和的nahco3溶液,调至ph=1,拌样,柱层析,展开剂为石油醚:乙酸乙酯=1:50,得到化合物(11)。
所得化合物(11)为黄色固体,收率为96%。m.p.34-35℃;
元素分析:实测值c%30.91h%2.23,计算值c%30.73h%2.06;
1hnmr(400mhz,dmso-d6,tms)δ(ppm):7.83–7.33(m,3h),6.57(d,j=4.1hz,1h),6.17(d,j=4.1hz,1h),2.20(s,3h);
ei-ms(70ev)m/z(%):388[m]+,186(90.88),227(39.22),172(100.0),272(14.30);
实施例12
向100ml单颈圆底烧瓶中,依次加入苯甲醛12mmol,丙二酸35mmol,吡啶12mmol,30ml的丙酮。加热150℃回流6-7h;向反应体系加入20ml水,随后用5%稀hcl调至ph=1,在调至的过程中,大量固体析出,抽滤,得化合物c12。
向100ml单颈圆底烧瓶中,依次加入12mmol化合物c12,nbs30mmol,四甲基胍2mmol,30ml冰醋酸,加热回流7-8h;加入饱和的nahco3溶液,调至ph=1,拌样,柱层析,展开剂为石油醚:乙酸乙酯=1:50,得到化合物(12)。
所得化合物(12)为黄色液体,收率为93%。n=1.5541;
元素分析:实测值c%37.17h%3.42,计算值c%37.30h%3.13;
1hnmr(400mhz,dmso-d6,tms)δ(ppm):7.38(d,j=36.6hz,5h),6.53(d,j=4.4hz,1h),6.12(d,j=4.4hz,1h),2.15(s,3h);
13cnmr(100mhz,dmso-d6,tms)δ(ppm):173.91,141.06,133.87,133.35,132.24,82.92,53.05,25.92;
elementalanalysisc10h10br2o2(c,h)(%):c,37.30;h,3.13;found:c,37.17;h,3.42;
实施例13
向100ml单颈圆底烧瓶中,加入溴对位取代苯甲醛8mmol,硝基甲烷25mmol,三乙胺8mmol,二氯甲烷20ml。150℃加热回流5-10h,tlc检测反应至结束。后处理,向反应体系加入约50ml水,二氯甲烷萃取,合并有机相,无水硫酸钠干燥;抽滤,拌样,乙酸乙酯和石油醚为洗脱剂,洗脱剂选用ea:pe=1:40,柱层析分离纯化得化合物h13。
向100ml单颈圆底烧瓶中,依次加入8mmol化合物h13,卤素单质8mmol,乙酸钠8mmol,15ml冰醋酸,tlc检测试验至反应结束;后处理,加入约30ml的饱和的硫代硫酸钠,除去过量的液溴;二氯甲烷萃取,合并有机相,无水硫酸钠干燥;抽滤,拌样,柱层析,洗脱剂为石油醚:乙酸乙酯=1:50,最终得到化合物(13)。
所得化合物(13)为黄色固体,收率为99%。m.p.83-85℃;
元素分析:实测值c%31.59h%1.93n%4.315计算值c%31.30h%1.64n%4.56;
ei-ms(70ev)m/z(%):305[m+],259.89(28.89),101.06(96.70),179.98(100.0),261.90(14.05);
实施例14
向100ml单颈圆底烧瓶中,加入烷氧基对位取代苯甲醛10mmol,硝基甲烷30mmol,乙酸铵12mmol,二氯甲烷30ml。150℃加热回流5-10h,tlc检测反应至结束。后处理,向反应体系加入约50ml水,二氯甲烷萃取,合并有机相,无水硫酸钠干燥;抽滤,拌样,乙酸乙酯和石油醚为洗脱剂,洗脱剂ea:pe=1:50,柱层析分离纯化得化合物h14。
向100ml单颈圆底烧瓶中,依次加入10mmol化合物h14,卤素单质10mmol,乙酸钠10mmol,20ml冰醋酸,tlc检测试验至反应结束;后处理,加入约30ml的饱和的硫代硫酸钠,除去过量的液溴;二氯甲烷萃取,合并有机相,无水硫酸钠干燥;抽滤,拌样,柱层析,洗脱剂为石油醚:乙酸乙酯=1:50,最终得到化合物(14)。
所得化合物(14)为黄色固体,收率为99%。m.p.64-65℃;
元素分析:实测值c%41.67h%3.23n%4.55计算值c%41.89h%3.12n%5.43;
1hnmr(400mhz,dmso-d6,tms)δ(ppm)8.77(s,1h),8.04(d,j=8.2hz,2h),7.11(d,j=28.2hz,2h),3.85(s,3h);
13cnmr(100mhz,dmso-d6,tms)δ(ppm)165.05,141.45,138.25,126.81,119.30,119.11,60.52;
ei-ms(70ev)m/z(%):257[m+],209.99(30.09),148.10(7.38),132.08(100.0),117.04(32.87);
实施例15
向100ml单颈圆底烧瓶中,加入烷氧基间位取代苯甲醛10mmol,硝基甲烷30mmol,乙酸钠10mmol,二氯甲烷30ml。150℃加热回流5-10h,tlc检测反应至结束。后处理,向反应体系加入约50ml水,二氯甲烷萃取,合并有机相,无水硫酸钠干燥;抽滤,拌样,乙酸乙酯和石油醚为洗脱剂,洗脱剂可以选用ea:pe=1:60,柱层析分离纯化得化合物h15。
向100ml单颈圆底烧瓶中,依次加入10mmol化合物h15,卤素单质10mmol,乙酸钠10mmol,20ml冰醋酸,tlc检测试验至反应结束;后处理,加入约30ml的饱和的硫代硫酸钠,除去过量的液溴;二氯甲烷萃取,合并有机相,无水硫酸钠干燥;抽滤,拌样,柱层析,洗脱剂为石油醚:乙酸乙酯=1:50,最终得到化合物(15)。
所得化合物(15)为黄色固体,收率为98%。m.p.70-71℃;
元素分析:实测值c%42.02h%3.37n%5.65计算值c%41.89h%3.12n%5.43;
ei-ms(70ev)m/z(%):257[m+],209.99(100.0),148.10(7.38),132.08(95.14),117.06(60.70);
实施例16
向100ml单颈圆底烧瓶中,加入氯对位取代苯甲醛10mmol,硝基甲烷30mmol,三乙胺10mmol,二氯甲烷20ml。150℃加热回流5-10h,tlc检测反应至结束。后处理,向反应体系加入约50ml水,二氯甲烷萃取,合并有机相,无水硫酸钠干燥;抽滤,拌样,乙酸乙酯和石油醚为洗脱剂,洗脱剂可以选用ea:pe=1:60,柱层析分离纯化得化合物h16。
向100ml单颈圆底烧瓶中,依次加入10mmol化合物h16,卤素单质10mmol,乙酸钠10mmol,20ml冰醋酸,tlc检测试验至反应结束;后处理,加入约30ml的饱和的硫代硫酸钠,除去过量的液溴;二氯甲烷萃取,合并有机相,无水硫酸钠干燥;抽滤,拌样,柱层析,洗脱剂为石油醚:乙酸乙酯=1:50,最终得到化合物(16)。
所得化合物(16)为黄色固体,收率为97%。m.p.90-92℃;
元素分析:实测值c%36.90h%2.17n%5.26计算值c%36.61h%1.92n%5.34;
1hnmr(400mhz,dmso-d6,tms)δ(ppm)8.79(s,1h),7.99(d,j=8.4hz,1h),7.61(d,j=8.5hz,2h);
13cnmr(100mhz,dmso-d6,tms)δ(ppm)141.20,140.39,137.20,133.89,133.56,130.55;
ei-ms(70ev)m/z(%):261[m]+,136.00(100.0),215.95(15.46),152.02(14.34);
实施例17
向100ml单颈圆底烧瓶中,加入氯邻位取代苯甲醛10mmol,硝基甲烷30mmol,三乙胺10mmol,二氯甲烷20ml。150℃加热回流5-10h,tlc检测反应至结束。后处理,向反应体系加入约50ml水,二氯甲烷萃取,合并有机相,无水硫酸钠干燥;抽滤,拌样,乙酸乙酯和石油醚为洗脱剂,洗脱剂可以选用ea:pe=1:45,柱层析分离纯化得化合物h17。
向100ml单颈圆底烧瓶中,依次加入10mmol化合物h17,卤素单质10mmol,乙酸钠10mmol,20ml冰醋酸,tlc检测试验至反应结束;后处理,加入约30ml的饱和的硫代硫酸钠,除去过量的液溴;二氯甲烷萃取,合并有机相,无水硫酸钠干燥;抽滤,拌样,柱层析,洗脱剂为石油醚:乙酸乙酯=1:50,最终得到化合物(17)。
所得化合物(17)为黄色液体,收率为98%。n=1.6163;
元素分析:实测值c%36.31h%2.17n%5.20计算值c%36.61h%1.92n%5.34;
实施例18
向100ml单颈圆底烧瓶中,加入苯甲醛10mmol,硝基甲烷30mmol,三乙胺10mmol,三氯甲烷20ml。150℃加热回流5-10h,tlc检测反应至结束。后处理,向反应体系加入约50ml水,二氯甲烷萃取,合并有机相,无水硫酸钠干燥;抽滤,拌样,乙酸乙酯和石油醚为洗脱剂,洗脱剂可以选用ea:pe=1:60,柱层析分离纯化得化合物h18。
向100ml单颈圆底烧瓶中,依次加入10mmol化合物h18,卤素单质10mmol,乙酸钠10mmol,20ml冰醋酸,tlc检测试验至反应结束;加入约30ml的饱和的硫代硫酸钠,除去过量的液溴;二氯甲烷萃取,合并有机相,无水硫酸钠干燥;抽滤,拌样,柱层析,洗脱剂为石油醚:乙酸乙酯=1:50,最终得到化合物(18)。
所得化合物(18)为黄色固体,收率为99%。m.p.63-64℃;
元素分析:实测值c%42.33h%2.93n%5.92计算值c%42.13h%2.65n%6.14;
13cnmr(100mhz,dmso-d6,tms)δ(ppm)141.52,136.58,135.57,134.93,133.62,132.81;
ei-ms(70ev)m/z(%):227[m+],102.04(100.0),183.90(53.74),104.00(6.68);
实施例19
向100ml单颈圆底烧瓶中,加入氯邻位和对位取代苯甲醛8mmol,硝基甲烷35mmol,乙酸30mmol,二氯甲烷20ml。150℃加热回流5-10h,tlc检测反应至结束。后处理,向反应体系加入约50ml水,二氯甲烷萃取,合并有机相,无水硫酸钠干燥;抽滤,拌样,乙酸乙酯和石油醚为洗脱剂,洗脱剂可以选用ea:pe=1:45,柱层析分离纯化得化合物h19。
向100ml单颈圆底烧瓶中,依次加入10mmol化合物h19,卤素单质10mmol,乙酸钠10mmol,20ml冰醋酸,tlc检测试验至反应结束;后处理,加入约30ml的饱和的硫代硫酸钠,除去过量的液溴;二氯甲烷萃取,合并有机相,无水硫酸钠干燥;抽滤,拌样,柱层析,洗脱剂为石油醚:乙酸乙酯=1:50,最终得到化合物(19)。
所得化合物(19)为黄色固体,收率为97%。m.p.61-63℃;
元素分析:实测值c%32.13h%1.06n%4.76计算值c%32.36h%1.36n%4.72。
ei-ms(70ev)m/z(%):295[m]+,169.95(100.0),261.88(31.59),185.86(6.35);
实施例20
向100ml单颈圆底烧瓶中,加入氯间位和对位取代苯甲醛12mmol,硝基甲烷30mmol,乙酸30mmol,二氯甲烷30ml。150℃加热回流5-10h,tlc检测反应至结束。后处理,向反应体系加入约50ml水,二氯甲烷萃取,合并有机相,无水硫酸钠干燥;抽滤,拌样,乙酸乙酯和石油醚为洗脱剂,洗脱剂可以选用ea:pe=1:45,柱层析分离纯化得化合物h20。
向100ml单颈圆底烧瓶中,依次加入10mmol化合物h20,卤素单质10mmol,乙酸钠10mmol,20ml冰醋酸,tlc检测试验至反应结束;后处理,加入约30ml的饱和的硫代硫酸钠,除去过量的液溴;二氯甲烷萃取,合并有机相,无水硫酸钠干燥;抽滤,拌样,柱层析,洗脱剂为石油醚:乙酸乙酯=1:50,最终得到化合物(20)。
所得化合物(20)为黄色固体,收率为96%。m.p.67-69℃;
元素分析:实测值c%32.36h%1.66n%4.65计算值c%32.36h%1.36n%4.72。
1hnmr(400mhz,dmso-d6,tms)δ(ppm)8.78(s,1h),8.19(s,1h),7.95(d,j=9.4hz,1h),7.81(d,j=8.5hz,1h);
13cnmr(100mhz,dmso-d6,tms)δ(ppm)143.14,139.55,138.80,137.60,136.15,135.54,134.26,132.52;
ei-ms(70ev)m/z(%):295[m]+,169.96(100.0),215.85(24.17),185.90(24.89);
实施例21
向100ml单颈圆底烧瓶中,加入磺酰基对位取代苯甲醛10mmol,硝基甲烷30mmol,三乙胺10mmol,二氯甲烷20ml。150℃加热回流5-10h,tlc检测反应至结束。后处理,向反应体系加入约50ml水,二氯甲烷萃取,合并有机相,无水硫酸钠干燥;抽滤,拌样,乙酸乙酯和石油醚为洗脱剂,洗脱剂可以选用ea:pe=1:45,柱层析分离纯化得化合物h21。
向100ml单颈圆底烧瓶中,依次加入10mmol化合物h21,卤素单质10mmol,乙酸钠10mmol,20ml冰醋酸,tlc检测试验至反应结束;后处理,加入约30ml的饱和的硫代硫酸钠,除去过量的液溴;二氯甲烷萃取,合并有机相,无水硫酸钠干燥;抽滤,拌样,柱层析,洗脱剂为石油醚:乙酸乙酯=1:50,最终得到化合物(21)。
所得化合物(21)为黄色固体,收率为99%。m.p.120-121℃;
元素分析:实测值c%35.25h%2.85n%4.43计算值c%35.31h%2.63n%4.51。
ei-ms(70ev)m/z(%):305[m]+,197.91(56.77),184.94(11.80);
实施例22杀虫活性测试
采用人工饲料表面涂布法,在96孔细胞培养板上测定了目标化合物对以上三种靶标的抑制活性。
表1杀虫活性的测试结果
表2复筛结果
可见,采用人工饲料表面涂布本申请的化合物,对小菜蛾、蚜虫、棉铃虫具有较高的致死率。
实施例23杀菌活性测试
采用离体平皿法,以测定菌丝的生长速率为依据来表征杀菌活性。配置药品溶液的过程如下:首先用分析天平称取目标化合物,用少许丙酮溶解,吐温-80乳化,最后加入蒸馏水配成一定浓度100μg/ml待用。
采用半固体培养法,以测定菌丝的生长速率为依据来表征杀菌活性。
表4化合物的杀菌活性2
由表可知,本申请的化合物对水稻稻瘟病菌、水稻纹枯病菌、小麦赤霉病菌、苹果轮纹病菌、灰霉病菌、桃褐腐病菌、夏季斑病菌、草坪币斑病菌、辣椒炭疽、辣椒疫霉、灰霉、小麦颖枯、番茄早疫、立枯和小麦赤霉具有较佳的杀菌活性。
实施例24水稻纹枯病防治实验
选取化合物(16)和化合物(17)代表的化合物进行田间小区实验。选择合适的试验地,种植感病品种糯优一号,于水稻纹枯病发病初期进行施药处理,药剂处理安排如下表5所示,其中井岗霉素和戊唑醇为对照药剂,清水为空白对照。每个处理小区面积30m2,不少于3次重复,区组随机排列。
表5药剂处理安排
药效结果调查:施药10天后检测药剂对水稻纹枯病的防治效果,每个小区按对角线5点取样,检测叶片的发病程度,并按照下面的分级方法,将叶片的发病情况进行分级和统计,计算病情指数和防治效果。
分级方法:
0级:无病;
1级:病斑面积占整片叶面积的5%以下;
3级:病斑面积占整片叶面积的6%~15%;
5级:病斑面积占整片叶面积的16%~25%;
7级:病斑面积占整片叶面积的26%~50%
9级:病斑面积占整片叶面积的50%以上。
病情指数的计算:
防治效果的计算:
化合物(16)和化合物(17)对水稻纹枯病(r.solani)的防治效果如图1所示,化合物(16)和(17)(60克/公顷,90克/公顷和120克/公顷)的药效要显著高于井冈霉素(90克/公顷)(p<0.05)。与戊唑醇相比,高剂量下(90克/公顷和120克/公顷),化合物(16)和(17)的药效要显著高于戊唑醇(75克/公顷)(p<0.05);低剂量下(60克/公顷),化合物(16)和(17)的药效高于戊唑醇(75克/公顷),但无显著性差异(p<0.05)。
综上所述,以化合物(16)和(17)为代表的肉桂酸衍生物农药对水稻纹枯病具有较好的防治效果,可进一步优化改进。
- 还没有人留言评论。精彩留言会获得点赞!