一种新型蒽醌衍生物及合成方法和应用与流程
本发明涉及化合物领域,具体涉及一种新型蒽醌衍生物及合成方法和应用。
背景技术:
铜是人体内排在铁、锌之后第三丰富的过渡金属元素。cu2+在人体内的适量存在有益于维持机体的正常工作。cu2+能参与体内酶反应、酶转录及一些氧化还原过程,同时还与人处于压力与恐慌下的生理反应密切相关。但是如果体内cu2+的代谢不正常,则可能会诱发一系列疾病,如menkes综合症、wilsom综合症、家族性肌萎缩症、阿尔茨海默氏症等。因此,设计并开发一种具有高灵敏度,高选择性检测cu2+的手段具有重要意义。
技术实现要素:
本发明是针对上述存在的技术问题提供一种新型蒽醌衍生物及合成方法和应用。
本发明的目的可以通过以下技术方案实现:
一种新型蒽醌衍生物,该蒽醌衍生物的结构如化合物iii所示:
一种上述新型蒽醌衍生物制备方法,其制备方法的反应路线如下:
在一些具体的技术方案中,该方法包括以下步骤:
第一步,2-氨基蒽醌与氯乙酰氯在碱性试剂存在的条件下进行反应,得到化合物ii;
第二步,化合物ii与间苯二胺在碱性试剂存在的条件下进行反应,得到化合物iii。
上述方法中:第一步反应中:反应溶剂为二氯甲烷、氯仿和四氢呋喃中的至少一种。
上述方法中:第一步反应是在碱性试剂条件下进行的,所示的碱性试剂为4-二甲氨基吡啶、吡啶和三乙胺中至少一种。
上述方法中:2-氨基蒽醌与氯乙酰氯的摩尔比为1:1~1.5,且2-氨基蒽醌与碱性试剂的摩尔比为1:1~10。
上述方法中:第二步反应中:化合物ii与间苯二胺的摩尔比为2~3:1,且间苯二胺与碱性试剂的摩尔比为1:1~5。
上述方法中:第二步反应中:反应溶剂为乙腈、二氯甲烷和乙醇中的至少一种;所示的碱性试剂为碘化钾、n,n-二异丙基乙胺、无水碳酸钾、4-二甲氨基吡啶、吡啶和三乙胺中至少一种。
本发明技术方案中:所述的蒽醌衍生物作为荧光化学传感器在检测cu2+中的应用。
本发明技术方案中:所述的蒽醌衍生物作为光化学传感器在检测cu2+中的应用。
本发明的有益效果:
本发明提供的蒽醌衍生物制备方法简单,且蒽醌衍生物作为荧光化学传感器灵敏度高。
附图说明
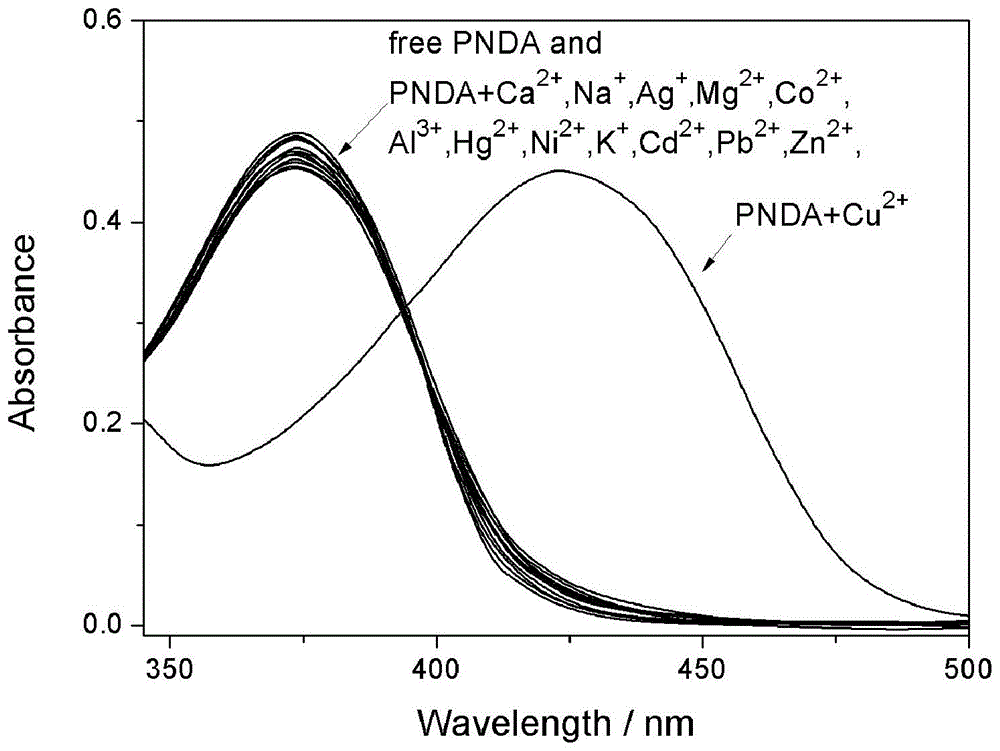
图1为探针分子pnda(实施例1)对cu2+的选择性吸收光谱识别。
图2为cu2+对探针分子pnda(实施例1)的吸收光谱滴定图。
图3是探针分子pnda(实施例1)对cu2+的选择性荧光光谱识别。
图4为cu2+对探针分子pnda(实施例1)的荧光光谱滴定图。
图5为cu2+与探针分子pnda(实施例1)反应时间对溶液荧光强度的影响图。
图6为当溶液中有其它共存金属离子时对探针pnda(实施例1)选择性识别cu2+的影响图。
图7为不同ph值对探针pnda(实施例1)选择性识别cu2+的影响图。
图8为探针pnda在不同cu2+浓度下与荧光强度的线性关系图。
具体实施方式
下面结合实施例对本发明做进一步说明,但本发明的保护范围不限于此:
实施例1
1、化合物ⅱ的合成
在500ml三颈烧瓶中依次加入2-氨基蒽醌(2.23g,10mmol)、150ml二氯甲烷和4-二甲氨基吡啶(1.22g,10mmol),在冰盐水浴中充分冷却,然后用恒压漏斗将50ml溶有氯乙酰氯(0.8ml,10mmol)的二氯甲烷溶液缓慢的滴加到充分搅拌的三颈烧瓶中,控制三颈烧瓶中的反应液温度不要超过0℃,滴加完成后,继续在冰盐水浴中反应6h。反应完成后,用0.1mol/l的naoh溶液调节反应液的ph值到9左右。然后用二氯甲烷(3×25ml)萃取反应液,合并有机相并用水(3×25ml)洗,再用无水na2so4干燥过夜。过滤后将滤液旋蒸,除去有机溶剂,得到产物ⅱ2.8g,产率:93.6%,纯度:99.36%。
元素分析:(%)forc16h10no3cl:计算值:c64.12;h3.36;n4.67,实测值:c64.87;h3.33;n4.59。
1hnmr(500mhz,cdcl3,tms):δ=10.41(s,1h),8.31(t,j=7.0,2h),8.16(s,1h),7.96-7.92(m,2h),7.84(d,j=7.2,2h),4.37(s,2h)ppm.
在250ml的烧瓶中,将间苯二胺(1.08g,10mmol)、碘化钾(3.32mg,0.02mmol)和n,n-二异丙基乙胺(20mmol)溶于100ml的乙腈中,在通入n2、回流和搅拌的条件下,用恒压漏斗缓慢滴加溶有化合物ⅱ(5.98g,20mmol)的50ml的乙腈溶液,控制在1h内滴完。滴加完成后继续回流反应20h,反应结束后将反应液冷却至室温,并将反应液倒入水中。然后用二氯甲烷(3×25ml)萃取,合并有机相,再用饱和nacl溶液(3×25ml)洗涤。有机相用无水na2so4干燥过夜。过滤后将滤液旋蒸,除去有机溶剂,得到产物ⅲ(pnda)5.88g,产率:92.7%,纯度:99.28%。
元素分析:(%)forc38h26n4o6:计算值:c71.92;h4.13;n8.83,实测值:c71.79;h4.08;n8.97。
1hnmr(500mhz,cdcl3,tms):δ=10.25(s,2h),8.30(t,j=7.2,4h),8.14(s,2h),7.94-7.89(m,4h),7.85(d,j=7.2,4h),7.05(t,j=7.0,1h),6.27(d,j=7.2,2h),5.77(s,1h),4.81(t,j=7.0,2h),3.92(d,j=7.2,4h)ppm.
实施例2
在250ml三颈烧瓶中依次加入2-氨基蒽醌(2.23g,10mmol)、150ml氯仿和吡啶(5ml,62mmol),在冰盐水浴中充分冷却,然后用恒压漏斗将50ml溶有氯乙酰氯(0.8ml,10mmol)的氯仿溶液缓慢的滴加到充分搅拌的三颈烧瓶中,控制三颈烧瓶中的反应液温度不要超过0℃,滴加完成后,继续在冰盐水浴中反应6h。反应完成后,用0.1mol/l的naoh溶液调节反应液的ph值到9左右。然后用二氯甲烷(3×25ml)萃取反应液,合并有机相并用水(3×25ml)洗,再用无水na2so4干燥过夜。过滤后将滤液旋蒸,除去有机溶剂,得到产物ⅱ2.73g,产率:91.3%,纯度:99.12%。
在250ml的烧瓶中,将间苯二胺(1.08g,10mmol)、碘化钾(3.32mg,0.02mmol)和三乙胺(20mmol)溶于100ml的二氯甲烷中,在通入n2、回流和搅拌的条件下,用恒压漏斗缓慢滴加溶有化合物ⅱ(5.98g,20mmol)的50ml的二氯甲烷溶液,控制在1h滴完。滴加完成后继续回流反应20h,反应结束后将反应液冷却至室温,并将反应液倒入水中。然后用二氯甲烷(3×25ml)萃取,合并有机相,再用饱和nacl溶液(3×25ml)洗涤。有机相用无水na2so4干燥过夜。过滤后将滤液旋蒸,除去有机溶剂,得到产物ⅲ(pnda)5.71g,产率:90.1%,纯度:99.21%。
实施例3
在250ml三颈烧瓶中依次加入2-氨基蒽醌(2.23g,10mmol)、150ml四氢呋喃和三乙胺(5ml,46mmol),在冰盐水浴中充分冷却,然后用恒压漏斗将50ml溶有氯乙酰氯(0.8ml,10mmol)的四氢呋喃溶液缓慢的滴加到充分搅拌的三颈烧瓶中,控制三颈烧瓶中的反应液温度不要超过0℃,滴加完成后,继续在冰盐水浴中反应6h。反应完成后,用0.1mol/l的naoh溶液调节反应液的ph值到9左右。然后用二氯甲烷(3×25ml)萃取反应液,合并有机相并用水(3×25ml)洗,再用无水na2so4干燥过夜。过滤后将滤液旋蒸,除去有机溶剂,得到产物ⅱ2.65g,产率:88.6%,纯度:99.15%。
在250ml的烧瓶中,将间苯二胺(1.08g,10mmol)、碘化钾(3.32mg,0.02mmol)和无水碳酸钾(20mmol)溶于100ml的乙醇中,在通入n2、回流和搅拌的条件下,用恒压漏斗缓慢滴加溶有化合物ⅱ(5.98g,20mmol)的50ml的乙醇溶液,控制在1h内滴完。滴加完成后继续回流反应20h,反应结束后将反应液冷却至室温,并将反应液倒入水中。然后用二氯甲烷(3×25ml)萃取,合并有机相,再用饱和nacl溶液(3×25ml)洗涤。有机相用无水na2so4干燥过夜。过滤后将滤液旋蒸,除去有机溶剂,得到产物ⅲ(pnda)5.62g,产率:88.6%,纯度:99.07%。
性质实验
1、吸收光谱实验
蒽醌衍生物pnda对cu2+的吸收光谱识别
图1是探针分子pnda(实施例1)对cu2+的选择性吸收光谱识别。在10ml浓度为0.1mmol/l探针分子pnda溶液中分别加入10μl浓度为0.1mol/l(1倍摩尔量)的金属离子溶液(ca2+、na+、ag+、mg2+、co2+、al3+、hg2+、ni2+、k+、cd2+、pb2+、zn2+、cu2+)。实验中所使用的溶液体系均为乙腈/水(3:1,v:v)的混合溶液,吸收光谱在岛津uv-2450型紫外分光光度计上测定。
由图1可以看出探针分子pnda(实施例1)在乙腈/水(3:1,v:v)的混合溶液中自身的吸收在373nm左右,当我们向探针分子溶液中加入过量的金属离子后,我们发现只有在加入cu2+后,溶液的吸收红移至425nm左右,溶液的颜色也由黄绿色变为橙黄色,而当在探针分子溶液中加入其它金属离子时,则没有这一现象的发生,这说明该探针分子的吸收光谱对cu2+有着独特的响应。
图2为cu2+对探针分子pnda(实施例1)的吸收光谱滴定图。在10ml浓度为0.1mmol/l探针pnda溶液中依次加入0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1.0、1.2、1.5、2.0倍摩尔量的cu2+。实验中所使用的溶液体系均为乙腈/水(3:1,v:v)的混合溶液,吸收光谱在岛津uv-2450型紫外分光光度计上测定。由图2可以看出,随着cu2+的加入,溶液的吸收波长逐渐由373nm红移至425nm,当cu2+加入量达到探针分子1倍摩尔量后,溶液的吸收波长不再移动,且峰的强度基本不变。这说明探针分子pnda与cu2+是1:1配位的。
2、荧光光谱实验
蒽醌衍生物pnda对cu2+的荧光识别
图3是探针分子pnda(实施例1)对cu2+的选择性荧光光谱识别。将探针分子pnda溶于乙腈/水(3:1,v:v)的混合溶液中,配制成浓度为10μmol/l的溶液,在此溶液中分别加入1倍摩尔量的金属离子(ca2+、na+、ag+、mg2+、co2+、al3+、hg2+、ni2+、k+、cd2+、pb2+、zn2+、cu2+)。激发波长为420nm,测定溶液的荧光光谱。从图3中可以看出,探针分子溶液仅在550nm处有一个很弱荧光发射峰,在加入cu2+后,溶液在490nm处出现了一个很强的荧光发射峰,而加入其它金属离子则没有这一现象,这说明该探针分子对cu2+表现出非常强的荧光选择识别性。实验中所使用的溶液体系均为乙腈/水(3:1,v:v)的混合溶液,荧光光谱在amincobowmanseries2荧光光谱仪上测得。
图4为cu2+对探针分子pnda(实施例1)的荧光光谱滴定图。在10μmol/l的探针分子pnda的乙腈/水(3:1,v:v)的混合溶液中,分别加入0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1.0、1.2、1.5、2.0倍摩尔量的cu2+。在420nm处激发,测量溶液的发射光谱,如图所示随着cu2+的浓度增加,在490nm处出现一个新的荧光发射峰,并且荧光发射峰的强度随着cu2+加入而不断增强,当cu2+的加入量达到1倍摩尔量探针分子后,490nm处的发射峰强度基本不再增加。
图5为cu2+与探针分子pnda(实施例1)反应时间对溶液荧光强度的影响图。在10μmol/l的探针分子pnda的乙腈/水(3:1,v:v)的混合溶液中,加入1倍摩尔量的cu2+。在激发波长420nm,发射波长490nm处,分别在0、0.5、1.0、1.5、2.0、2.5、3.0、3.5、4.0、4.5、5.0分钟时记录溶液的荧光强度。如图所示,在探针分子pnda溶液中加入cu2+2分钟后,荧光强度达到最大值,且随着时间延长基本保持不变。
图6为当溶液中有其它共存金属离子时对探针pnda(实施例1)选择性识别cu2+的影响图。在10μmol/l的探针分子pnda的乙腈/水(3:1,v:v)的混合溶液中,分别加入溶有10倍摩尔量的金属离子(ca2+、na+、ag+、mg2+、co2+、al3+、hg2+、ni2+、k+、cd2+、pb2+、zn2+),在激发波长420nm,发射波长490nm处,测量溶液的荧光强度,然后再在上述溶液中加入1倍摩尔量的cu2+,在激发波长420nm,发射波长490nm处,测量溶液的荧光强度,从图6中可以看出,当溶液中大量存在其他金属离子时,探针分子pnda对cu2+的选择性识别并不受影响。
图7为不同ph值对探针pnda(实施例1)选择性识别cu2+的影响图。分别用不同浓度的盐酸或氢氧化钠溶液以调节10μmol/l的探针分子pnda的乙腈/水(3:1,v:v)的混合溶液的ph值,在激发波长420nm,发射波长490nm条件下,测量探针溶液的荧光强度;然后再在以上溶液中分别加入1倍摩尔量的cu2+,在激发波长420nm,发射波长490nm条件下,测量溶液的荧光强度。从图7中可以看出,在ph=5-10的范围内探针分子对cu2+都具有很好荧光响应,并且比较稳定,这说明该探针可以在更宽泛的环境中检测cu2+。
图8为探针pnda(实施例1)在不同cu2+浓度与荧光强度的线性关系图。从图中可以看出当cu2+浓度在0.05-0.6mmol/l范围内呈现出良好的线性关系(r2=0.9983),纵坐标i为探针溶液中加入cu2+后所测得的荧光强度,i0为探针溶液中未加入cu2+后所测得的荧光强度,使用3σiupac标准计算所得的检测限为2.37×10-7mol/l。
- 还没有人留言评论。精彩留言会获得点赞!