用于制备乙二醇的方法与流程
相关申请的交叉引用本发明要求于2018年4月30日提交的美国临时专利申请no.62/664,603的权益,其全部内容和公开内容通过引用合并于此。
背景技术:
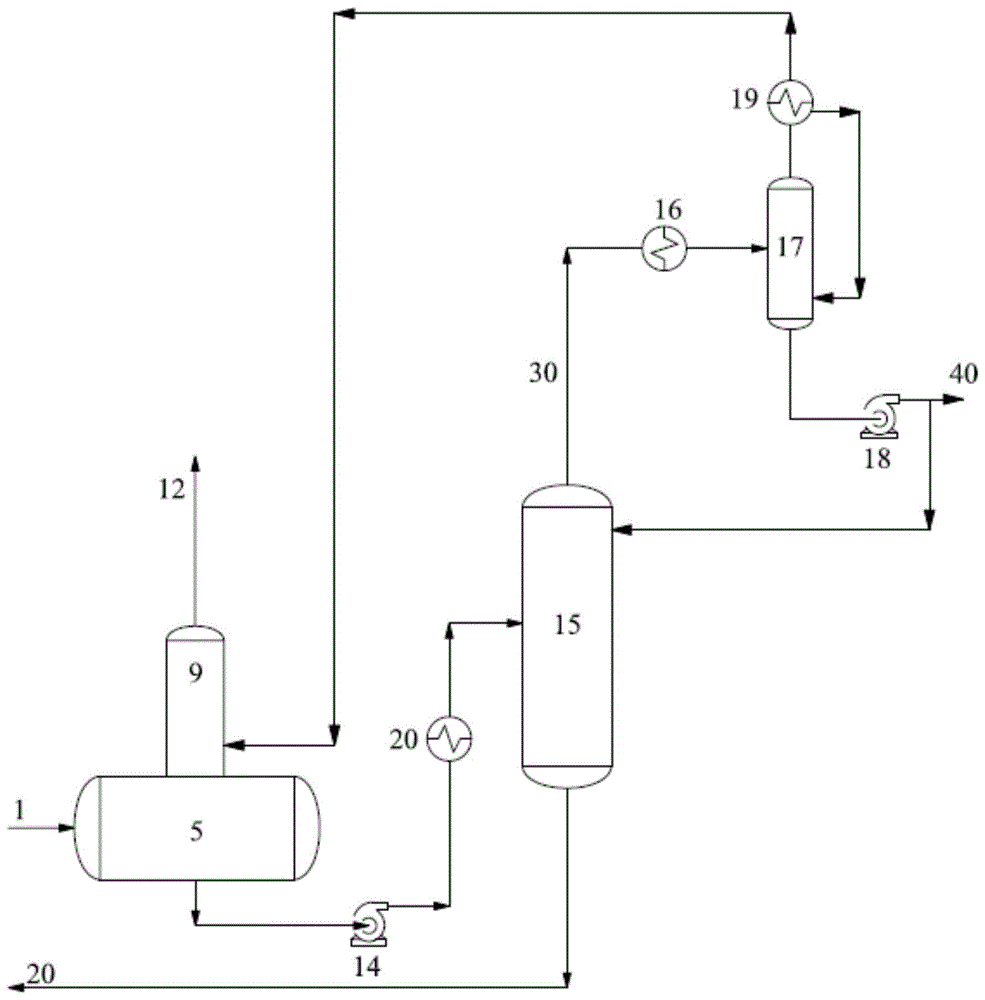
:乙二醇在社会中的作用从其第一次合成后不久的时间已经大大提高,该分子的性质成为两个早期有机化学巨头adolphewurtz和hermannkolbe之间激烈争议的主题。特别是,wurtz和kolbe争议在有关酒精同系物的新认识中乙二醇的功能和化学式,这对于kolbe关于化学结构的更多理论至关重要。该实验室很快就成为了德国和法国之间竞争日益激烈的工业和技术力量的代理战争,两国都投入了大量资源进行科学调查,而wurtz在化学合成方面的魔力赋予法国相当大的优势。该争议最终仅仅凭借武力在俾斯麦的德国联邦因法国-普鲁士战争而吞并了wurtz的阿尔萨斯家园时终结,并且因此使国际争议本质上变成了国内争议。今天,对乙二醇的兴趣更加平和,但更具竞争力,因为乙二醇是最广泛生产的有机化学品之一。由于乙二醇的大规模工业生产始于第一次世界大战前夕,因此为汽车和其他车辆提供动力的内燃机的使用的急剧增加刺激了对乙二醇作为冷却剂和防冻剂的需求。从那时起,乙二醇产量的增加只会加速,以至于到2017年,全球乙二醇产量估计超过250亿吨。通常将乙二醇制备为环氧乙烷的许多衍生物中的一种,并且尽管可使用其他生产途径,但大多数是在液相非催化热水合方法中由环氧乙烷制备的。因为环氧乙烷与乙二醇反应比与水反应更容易,所以不可避免地会形成单乙二醇(meg)和高级乙二醇副产物的混合物,高级乙二醇副产物如二甘醇(deg)、三甘醇(teg)、以及甚至更高级的乙二醇。虽然这些较高级的甘醇具有相当大的经济价值,但许多生产商和工厂经营者希望避免生产它们,因为这些产品的最终用户市场不是很发达,并且可能难以找到并将这些高级甘醇分配给对其有需求的工业用户。为了抑制产物甘醇和环氧乙烷之间的反应并且因此减少这些高级甘醇的形成,常规的非催化水合用一定量的水进行,其中水的量远远超过用于将环氧乙烷水合为乙二醇的化学计算量的水,例如每摩尔环氧乙烷15至40摩尔水。这种过量水的添加有效地平衡了产物甘醇和环氧乙烷之间的动力学上有利的竞争反应,如上所述,其与将环氧乙烷转变为单乙二醇的水合作用相竞争。然而,尽管有效地抑制了较高乙二醇的产生,但是相对于环氧乙烷使用大量过量的水对于工厂操作者来说存在去除这些大量过量的未反应水的问题,因为这种去除是能量密集的并且需要大规模的蒸发/蒸馏设施。因此,已经对用于生产乙二醇的环氧乙烷的标准热水合作用的替代物进行了深入研究。常规热水合的替代方案的一个早期示例是环氧乙烷均匀催化水合成单乙二醇。该方式的最早示例包括硫酸及其相关盐的均相催化(参见othmer,d.f.andthakar,m.s.,glycolproduction-hydrationofethyleneoxide.ind.eng.chem.1958,50,1235)。欧洲专利no.0123700描述了这些前几代酸催化剂的改进,其中用例如乙胺处理它们以部分中和它们,以期改善水合反应对单乙二醇的选择性。从那时起,已经提出了其他盐用于均相体系,例如美国专利no.4,160,116中所述的季鏻盐和美国专利no.7,683,221中所述的金属盐和碳酸氢盐。还提出了越来越有创意的有机物质(如edta和salen化合物)的组合作为均相催化剂(参见hal,j.w.,ledford,j.s.,andzhang,x.,catalysistoday123(2007),310-315)。均相催化剂体系通常用于制备乙二醇的两步法中,参见例如美国专利no.4,519,875,其中环氧乙烷首先与二氧化碳反应以制备碳酸亚乙酯,然后将其水解成乙二醇,通常在两个步骤中使用相同的催化剂。在这项开创性的专利之后,持续的研究已经在两步过程中产生了渐进的改进。例如,在美国专利no.5,763,691中,碳酸化反应在卤代有机鏻盐碳酸化催化剂存在下在环氧乙烷吸收物中催化。其他研究大大扩展了已知催化剂的范围;参见例如美国专利no.7,453,015中描述的大环螯合化合物(“冠醚”)。虽然均相催化剂与非催化水合相比改善了水合反应对乙二醇的选择性,但均相催化剂具有不足以抵消这些改进的缺点。特别地,甘醇产物溶液含有可溶或悬浮的均相催化剂-基本上该催化剂污染了产物,该催化剂对产品质量有害并且具有高腐蚀性,例如在酸催化剂的情况下。因此,需要将均相催化剂与甘醇产物溶液分离的额外步骤,这增加了该方法的成本和复杂性。例如,虽然上述美国专利no.5,763,691试图通过使环氧乙烷和二氧化碳在吸收物中反应来简化两步乙二醇的制备,但该专利仍然需要单独的蒸馏步骤来除去碳酸化催化剂。该方法复杂性带来了额外的缺陷,即它既不是非常通用也不是灵活的。例如,该方法缺乏部分催化操作的灵活性,即,在混合催化和非催化模式下操作的灵活性-如果操作者希望的话,这将允许产生更高级的甘醇,并且在某些后勤或操作紊乱情况下也可以适应设施,在这种紊乱情况下,例如当没有足够的催化剂时,其无法以催化模式运行。两步水合方法也缺乏用于新设施和改造的多功能性。两步水化方法只能用于新设施,不能用于改造,因为在改造的情况下,需要拆除和更换整个“后端”,这使得这种改造成本过高。两步水合方法的缺点是需要在该方法的第一步中存储浓缩的环氧乙烷(浓度大于70%)以生产碳酸亚乙酯中间体。第一步中的碳酸化反应是通过在高温高压(例如,大于1.6mpa)下将二氧化碳鼓泡到该浓缩的液体环氧乙烷溶液中进行的。大量浓缩的环氧乙烷需要相当多的额外措施(例如,提供液体冷藏)以防止可能导致任何气流(尤其是含有浓度高于典型的环氧乙烷的浓度的那些气流)的点燃、燃烧、爆燃、爆破或爆炸的反应或事件。不出所料,考虑到非催化和均相催化水合体系的上述缺点,还研究了使用固体或负载型催化剂的非均相催化。那些寻求具有较高选择性的催化水合作用而没有上述缺点的人已将他们的注意力转向非均相催化剂体系。因为非均相催化剂不被吸收到液相反应或产物溶液中,不会污染产物溶液,不需要分离步骤,并且便于水合反应器的连续操作,因此对它们有相当大的兴趣。已经提出多种固体材料作为用于水合的非均相催化剂。该清单包括沸石,如美国专利no.3,028,434所述的沸石,以及无定形硅铝酸盐,固体碱和负载型酸催化剂,如美国专利no.4,165,440所公开的那些。还提出了离子交换树脂,其具有描述于ep0123709中的部分胺中和的磺酸苯乙烯-二乙烯基苯树脂;季鏻离子交换树脂催化剂,参见pct公开wo2000/35840;美国专利no.6,211,419总结的强碱性离子交换树脂,其属于被提出用于催化水和环氧乙烷反应的第一批离子交换树脂。最近的出版物已将该领域扩展到宽幅阴离子交换树脂,参见例如中国专利no.1208135。虽然已经使用非均相离子交换树脂催化剂获得了很好的结果,但是它们的使用也存在某些缺点。特别地,离子交换树脂具有有限的耐热性,使得在典型反应条件下经历长的使用时间,树脂经历溶胀,导致失活。由树脂床内的内部聚合和高分子量副产物的总体积聚引起的溶胀是一个严重的问题,它是离子交换树脂寿命的主要限制因素。树脂在操作过程中因为它们暴露在溶剂中而通常会经历一些可逆的溶胀;然而,通过仔细管理,可以减轻这种溶胀并延长离子交换树脂的有效寿命。然而,离子交换树脂暴露于环氧乙烷会导致树脂更加的问题和不可逆的溶胀-特别是当在水合反应器的较高温度下发生这种暴露时,如上所述,该温度可能超出树脂可以令人满意地耐受的温度范围。溶胀不仅本身是一个问题,而且还会降低效率并使催化水合方法复杂化;例如,当过量溶胀需要更换树脂,并且因此含有它的催化水合单元脱机时。已经进行了各种尝试来解决这些困难,包括尝试在典型的操作条件下找到不易于溶胀的树脂,并且另外开发各种方法改进方案以试图改善性能以及减少溶胀。例如,ep专利no.1828086公开了使用设计用于去除据称可导致树脂溶胀的杂质的“保护床”,但这种努力是无效的。不是用保护床保护树脂以试图使溶胀最小化,pct公开wo2008/150338中概述的另一种更成功的方法寻求不保护树脂,而是促进通过迄今为止未建议的以上流模式操作催化反应器来去除超过使用寿命的离子交换树脂的方法,其使催化剂床溶胀,允许催化剂颗粒按尺寸分离,从而选择性地除去溶胀颗粒。另一种方式是美国专利no.6,160,187中所示的,其声称在绝热反应条件下和使用绝热反应器可以实现溶胀的减少。然而,no.6,160,187专利中包含的实施例实际上没有证明树脂的溶胀性能有任何改善,也没有随后的绝热操作的发展,其发展已被忽略。因此,尽管在非均相催化水合的各个方面存在若干有希望的发展,但仍然非常需要在绝热方法条件下进一步改进催化水合作用。技术实现要素:本发明涉及一种用于生产乙二醇的液相方法,其包括:提供水流和含水环氧乙烷进料流;将所述含水环氧乙烷进料流分成至少相等的第一部分和相等的第二部分;将所述第一部分和所述水流合并以形成第一进料流;将所述第一进料流进给至第一绝热反应器的入口内,所述第一绝热反应器的所述入口处于入口温度;使所述环氧乙烷和水在第一绝热反应器中在第一离子交换树脂催化剂存在下反应,从而产生含有水、乙二醇和未反应的环氧乙烷的第一反应器流出物流;从所述第一绝热反应器的出口排出所述第一反应器流出物流;使通过位于所述第一绝热反应器下游的热交换器的所述第一反应器流出物流冷却至水合温度;以及将所述第二部分和冷却的产物流合并以形成第二进料流;将所述第二进料流输送到第二绝热反应器的入口,所述第二绝热反应器的所述入口处于水解温度;在所述第二绝热反应器中在第二离子交换树脂催化剂存在下使所述第二进料流中所包含的环氧乙烷和水反应,从而产生含有水、乙二醇和未反应的环氧乙烷的第二反应器流出物流,其中所述第一绝热反应器和所述第二绝热反应器各自在固定催化剂床中含有离子交换树脂催化剂,并且所述出口温度高于所述入口温度。附图说明本发明的前面的概述以及以下优选实施方案的详细描述在结合附图一起阅读时将被更好地理解。为了说明本发明的目的,在附图中示出了目前优选的实施方案。然而,应该理解的是,本发明不限于所示的精确配置和工具。在附图中:图1是用于制备本发明包含的浓缩环氧乙烷流的方法的示意流程图;图2-3是根据本发明制备乙二醇的方法实施方案的示意流程图;以及图4是显示根据本发明的用于制备乙二醇的模拟示例性方法的示意流程图。具体实施方式本文所用的所有部分、百分数和比率都用体积表示,除非另有规定。所有压强都是绝对的。本文引用的所有文献都通过引用并入。所谓“水”是指任何类型的适合于在化学和石油化学处理中使用的水,包括去离子水、软化水、工业水、饮用水和蒸馏水。也可以使用从甘醇制造方法的脱水和蒸发部分获得的蒸气冷凝物和冷凝物。通过本发明,已经开发了一种改进的绝热催化水合方法,其中催化剂溶胀减少,方法灵活性显著提高,并且该方法配置成使得它可以有利地用于将非催化方法改造为催化方法。本发明的一个特别的优点是使用一系列绝热反应器,向每个绝热反应器注入新鲜的环氧乙烷水溶液。通常,非催化水合反应在单个绝热反应器中进行,而不考虑反应器的出口或最高温度。这种操作可能对本发明的离子交换树脂造成特别的危害,因为如上所述,在指定用于这种树脂的温度范围之外的温度会增加溶胀并降低催化剂寿命。本发明不是使用这些单级绝热反应器和伴随它们的缺点(例如在催化剂存在下的高温),而是使用串联布置的多级绝热反应器,其中对每一级进行外部级间冷却并将新鲜的环氧乙烷水溶液注入每一级。由于在绝热反应器中没有除去热量,所以流出物的温度超过进料的温度,并且必须在每一级之后通过冷却剂从流出物中除去反应热。这对于防止上述树脂溶胀并防止产物甘醇与环氧乙烷过度非催化反应而形成高级甘醇特别重要。此外,常规的非催化绝热反应器不能有利地配置成具有串联的其中注入新鲜的环氧乙烷水溶液的多个反应器级。在这种配置中,将新鲜的环氧乙烷引入含有可观浓度的产物甘醇的反应器流出物流中会导致产物甘醇过量反应成不希望有的高级甘醇。在本发明中,通过提供催化水合反应部分来解决这些问题,所述催化水合反应部分具有串联的两个或更多个绝热反应器,每个反应器之间有级间冷却,以及向每个反应器注入新鲜的水性环氧乙烷溶液。级间冷却除去流出物的反应热,降低其温度,使其可以与新鲜的环氧乙烷水溶液合并用于下一个连续的反应器级。本发明利用来自每个级间冷却器的冷却流出物温度和注入每个反应器的新鲜水性环氧乙烷的比例,以将每个反应器中的最高温度控制在催化剂寿命和性能所需的范围内。此外,从工艺角度来看,通过外部级间热交换器进行冷却比配置适当的内部反应器冷却负荷更容易、更有效和更简单,并且可以调节以吸收方法中的正常波动,同时保持催化反应器的性能。因此,如本发明使用一系列绝热反应器改进了方法的多功能性,并使得能将常规的非催化水合方法改造或改装成催化方法,这将在下面更详细地描述。在本发明中,实现了这些益处,同时仍然保留了先前催化水合体系对单乙二醇的高选择性。在每个绝热反应器床中,环氧乙烷和水在非均相催化剂(如离子交换树脂)存在下反应以形成单乙二醇,其中至少98%的环氧乙烷转化为单乙二醇,只有很小的百分比被转化进入更高级的甘醇,如二甘醇或三甘醇。因此,通过本发明,已经开发了一种改进的方法方案,其包括含有串联的绝热反应器的催化水合反应部分,其保持低树脂溶胀和优异的选择性,同时还降低了方法复杂性并增加了多功能性。如上所述根据本发明制备的该催化水合反应部分(也称为“反应部分”)在图2中显示为25,并将在下面更详细地描述。除了其他进料流之外,还用水进料流36进给反应部分25,用液体冷进料流导管40进给反应部分25,液体冷进料流导管40高度富含环氧乙烷并且在足够高的压强下提供以使环氧乙烷保持液态。反应部分25包括精加工绝热非催化热eo水解反应器26,其确保剩余的未转化的eo(存在于反应器序列22的最后绝热反应器流出物中)完全转化为ppm水平,从而显著减少所需的反应器序列22的催化反应器总体积。用于产生高度富含环氧乙烷的该液体进料流的任何合适的装置适用于本发明的用于催化生产乙二醇的方法。用于这样做的一种合适的装置是图1中所示的直接两级闪蒸吸收器-汽提塔部分。图2显示了吸收器-汽提塔部分1和已根据本发明制备的反应部分25之间的连通。现在将更详细地描述参考该直接两级闪蒸吸收器-汽提塔部分制备环氧乙烷的方法。环氧乙烷是在环氧乙烷(“环氧化”)催化剂(在下面更详细描述)的存在下通过使含氧气体与烯烃(优选乙烯)连续地接触来生产的。氧气可以以基本上纯分子形式或以混合物(如空气)的形式供给到反应中。举例而言,在操作条件下典型的反应物进料混合物可以包含约0.5%至约45%(优选约5%至约30%)的乙烯,约3%至约15%的氧气,余量包含相对惰性的材料,所述惰性的材料包括诸如二氧化碳、水、惰性气体、其他烃类、以及本文所描述的反应缓和剂等这样的物质。惰性气体的非限制性示例包括氮气、氩气、氦气以及它们的混合物。其他烃类的非限制性示例包括甲烷、乙烷、丙烷和它们的混合物。二氧化碳和水是环氧化方法的副产物以及进料气体中的普通污染物。它们都对催化剂有不良影响,因此,这些组分的浓度通常保持在最小量。如前面所提到的,在反应中还存在一种或多种反应缓和剂。其非限制性的示例包括有机含卤素化合物,如c1至c8卤代烃;特别优选的是含氯化物缓和剂,如氯甲烷、氯乙烷、二氯乙烷、氯乙烯或它们的混合物。控制氯化物浓度水平对含铼催化剂而言特别重要。如上所述,用于乙烯环氧化方法的常规方法包括在固定床管式反应器中在环氧化催化剂存在下利用分子氧气相氧化乙烯。常规的商业固定床乙烯-氧化物反应器通常为多个平行细长管(在合适的壳体中)的形式,约0.7至2.7英寸o.d.和0.5至2.5英寸i.d.和15-53英尺长,每个填充和封装有催化剂。将反应进料混合物(如上所述)引入这些管中,并且所得反应器流出物气体含有环氧乙烷、未使用的反应物和副产物。仅出于说明的目的,以下是当前商业环氧乙烷反应器单元中经常使用的条件:气时空速(ghsv)为1500-10,000h-1,反应器入口压强为150-400psig,冷却剂温度为180-315℃,氧气转化率为10-60%,以及eo生成率(工作率)为7-20磅eo/立方英尺催化剂/小时。在启动完成和正常操作期间反应器入口中的进料组成通常包含(按体积%计)1-40%乙烯,3-12%o2;0.3%至20%,优选0.3至5%,更优选0.3至1%co2;0-3%乙烷,一种或多种氯化物缓和剂的本文描述的量;以及进料的余量,其包含氩气、甲烷、氮气或其混合物。乙烯环氧化方法的典型操作条件包括范围为约180℃至约330℃,优选约200℃至约325℃,更优选约225℃至约280℃的温度。操作压强可以从约大气压到约30个大气压变化,具体取决于所需的质量速度和生产率。在本发明的范围内可以采用更高的压强。商业规模反应器中的停留时间通常为约2至约20秒。在图1中,洗涤塔液体(下文中称为“富循环水”流)流过导管1并供应到闪蒸罐5。富循环水流是包含1.5mol%至4mol%的环氧乙烷与一些溶解的轻质气体如甲烷、乙烯等的含水流。该富循环水流以优选在约0.4mpa至约0.6mpa之间的压强供应至闪蒸罐5。吸收器单元9永久并且直接且永久地固定至闪蒸罐5的顶部,并在闪蒸罐中提供开口以与吸收器9连通。在本发明中,闪蒸罐5和吸收器9组件中的压强保持在足够的范围内,使得当富循环水进入闪蒸罐5时“轻质”气体(其溶解在富循环水中,挥发性大于环氧乙烷)在进入闪蒸罐5时从富循环水中闪蒸出来,并且轻质气体蒸气由闪蒸罐5和吸收器9之间的压差引导向上通过开口进入吸收器9以形成吸收器9的顶部流出物,而环氧乙烷溶质很大程度地保持溶解在闪蒸罐5的液体底部中的富循环水流中。这些更易挥发的“轻质”气体包括上述惰性、未反应或副产物富含气体的循环水溶质(此外,微量环氧乙烷也可能与富循环水流中挥发性较强的轻质气体一起蒸发)。通过向下移动的洗涤流(未示出)以吸收和回收向上移动的环氧乙烷,可以回收从富循环水中起泡并与轻质气体溶质蒸气一起上升到吸收器9中的少量环氧乙烷。如上所述,吸收器9通过例如焊接直接且永久地固定到闪蒸罐5的顶表面上。吸收器9以与本领域技术人员公知的静柱和再沸器配置相同的方式固定到闪蒸罐5上。更具体地,吸收器9沿着闪蒸罐5的纵向轴线固定到闪蒸罐5上,但是优选地固定在纵向轴线的中点处。吸收器9的横截面与闪蒸罐5中的开口的形状相匹配,吸收器9固定在该开口上,并且该开口使得闪蒸罐5和吸收器9之间能连通。吸收器的横截面和闪蒸罐5中的开口的形状具有相同的匹配形状,并且该形状可以是圆形和椭圆形之间的任何形状,在偏心率为0和偏心率为1之间。闪蒸罐5中的开口和吸收器9是对准的,使得开口的中心与吸收器9的中心共线。配置闪蒸罐5中的开口和吸收器9的形状以及它们相应的对准确保了在这两个元件之间存在适当的流体连通。吸收器9优选地与闪蒸罐5中形成的开口的内径齐平地焊接,使得吸收器9的外径与闪蒸罐5中的开口的直径齐平。为了确保闪蒸更易挥发的“轻质”溶质同时保持环氧乙烷在富循环水中的高溶解度之间的平衡,闪蒸罐5中的压强保持在约0.1mpa至约0.3mpa,并且与其直接连通的吸收器9中的压强略低于闪蒸罐5中的压强。吸收器9顶部的气体温度为约30℃至约45℃,而闪蒸罐5中的温度为约50℃至约60℃。吸收器9的顶部流出物组分通过导管12返回上游反应部分(未示出),尽可能循环到反应器。富循环水作为液体底部从闪蒸罐5流动通过泵14以及通过可选的热交换器20(其预热流),并到达汽提塔15。汽提塔15中的压强通过控制由从排放冷凝器19排出的蒸气提供的背压来维持。汽提塔15顶部的温度为约25℃至约40℃,汽提塔15底部温度为约120℃至约160℃。富循环水流可以在热交换器20中预热,以减少操作汽提塔15所需的负荷。汽提塔15中的压强保持在约0.2mpa至约0.6mpa的范围内。汽提塔15还可以使用本领域技术人员公知的蒸气喷射器系统,其中蒸气由(1)在环氧乙烷生产设施中的其他地方产生的蒸气或(2)外部供应的中压或高压蒸气/osbl;或这两种源的组合供应。如图2所示,加压汽提塔塔顶流出物流通过导管30流到反应部分25。该流流入冷凝器16,然后流到闪蒸罐17,闪蒸罐17将闪蒸的轻质气体与进给泵18的浓缩的环氧乙烷水溶液流分离。泵18的排出物分成两股流:一股流作为回流物返回汽提塔15,一股净产物液体含水流浓缩在环氧乙烷中。来自闪蒸罐17的闪蒸轻质气体流到排放冷凝器19,以进一步从闪蒸蒸气流中回收环氧乙烷,并控制浓缩液体环氧乙烷溶液40中溶解的轻质气体的量。与在常规方法的环氧乙烷回收部分中相当的位置中发现的高度稀释的水溶液相比,加压汽提塔塔顶流出物流在环氧乙烷中富含更多,含有至少约40mol%,优选至少约50mol%的环氧乙烷,余量大部分为水。因此,加压的汽提塔塔顶流出物流以高浓度的环氧乙烷和高压供应。这使得该流非常适用于通过催化水解在非常经济的基础上进一步加工成乙二醇,因为该流不需要在下游加工步骤中进一步富集或加压。本发明的另一有利的方法选择是汽提塔15的液体底部物可以在优选的实施方案中,在设施中的其他地方,在各种其他热集成方案中使用,具体取决于需要和条件,该液体底部物是含有少量环氧乙烷(约1摩尔ppm至约50摩尔ppm)的贫循环水溶液,并且也不含轻质组分和杂质。在流相比于典型的环氧乙烷的浓度具有较高的浓度的这些情况下,操作者将继续保持在生产、处理或储存环氧乙烷时总是观察到的适当的安全标准。一如既往,必须采取措施以防止可能导致任何气流(尤其是含有浓度高于典型的环氧乙烷的浓度的那些气流)的点燃、燃烧、爆燃、爆破或爆炸的反应或事件。因此,为了防止这种情况,可以使用减压装置来减轻或减少在方法、反应或分离系统或在所示位置的上游和下游的环氧乙烷装置中的其他地方积聚的不希望有的压强。汽提塔15中的分离是如此有效,使得进入汽提塔15的富循环水中的几乎所有环氧乙烷成功地从富循环水中分离和回收并且作为净液体塔顶流出物流离开汽提塔15,而仅仅非常小部分的eo在别处离开汽提塔15。在汽提塔15中制备的加压汽提塔塔顶流出物流在高压下通过导管30从汽提塔部分1流到反应部分25,如图2所示。在本发明中,反应部分含有两个或多个反应器。在图2中,示出了反应部分25,其具有串联布置的一系列四个催化反应器,每个催化反应器通过热交换器分开。具体地,如在图2中示出的并且还在图3中更详细地示出的,反应器部分25示出了反应器序列22,反应器序列22具有四个向下流动式的、固定床的、串联布置的绝热反应器38、40、46和52与三个级间冷却器,即在线热交换器39、43和49。反应器被指定为第一绝热反应器38、第二绝热反应器40、第三绝热反应器46和第四绝热反应器52,但是在本文中使用的这些绝热反应器38、40、46和52也可称为“反应器”。如上所述,这种级间冷却是必要的,因为水与环氧乙烷的反应是高度放热的,因此当反应如在本发明中一样在绝热反应器中进行时,必须从产物或出口流中除去反应热,以避免会损害温度敏感催化剂(如离子交换树脂)的寿命的反应器床的过度的温度升高。这不同于直接从反应器中除去反应热的等温条件。在树脂能够耐受的温度下维持离子交换树脂催化剂对于减少溶胀、防止降解和最大化催化剂寿命当然是至关重要的。如果操作正确,可以将绝热温度升高控制到保持良好性能和寿命的水平。最后,位于反应器序列22下游的是传统的非催化绝热管式反应器26(该反应器当然不含催化剂)。当加压汽提塔塔顶流出物流流过导管30进入反应器部分时,它在四个串联的反应器中被分成相等的部分;然后将第一相等的部分的高度浓缩的环氧乙烷流与水源36(预先在热交换器35中加热)在混合装置(例如静态混合器)中混合,以产生稀释的含水环氧乙烷进料流37。流36在热交换器35中加热,以使混合流37达到介于50℃和90℃之间的温度。还存在水流36。水流36可以取自任何水源,包括osbl,但优选取自环氧乙烷/乙二醇设施中的另一部分。例如,水可以来自回收的冷凝水,该冷凝水来自设施的乙二醇蒸发部分-具体地,水可以从蒸发部分中的一个或多个蒸发器的顶部蒸气冷凝得到。水流36和流过管道30的加压汽提塔塔顶流出物流当会合在一起时形成流向反应器序列22的合并的稀释进料流。与环氧乙烷水合成乙二醇所需要的水量相比,该合并的进料流将具有化学计量过量的水(相对于环氧乙烷)。然而,该过量的量远远低于现有技术中使用的过量的量。因此,在本发明中,合并的流40和36具有约5:1至约15:1,优选约7:1至约12:1的水:环氧乙烷的摩尔比。因此,由于合并的总进料流含有较少的水,所以最终的反应器序列产物流88也将具有相应较少量的需要从产物中除去的水,如下所述。将汽提塔产物液体流40中所含的含水环氧乙烷分成四份大致相等的流57、61、64和67,这些流中的每一个含有大约相等摩尔量的水和环氧乙烷。其中,将流57与水流36(在热交换器35中加热后)合并,形成流向反应器38的进料流37,使得进料流37中水:环氧乙烷的摩尔比为约40:1至约10:1,优选约30:1至约20:1。这是对催化和非催化水合的常规操作的显著改进。通过将流57与水流36共同进给到第一反应器38并且然后将含水环氧乙烷进料流40的其他相等部分(61、64、67)与反应器流出物(70、73、76)共同进给,可以获得在流向每个反应器的进料流中的高稀释水:环氧乙烷比率,然而同时在反应器部分末端的被供给蒸发器进行水去除的产物流88中保持较低的全部水:环氧乙烷的比率。因此,环氧乙烷和水的反应在离子交换树脂存在下且在显著过量的水(相对于环氧乙烷)下被催化。这促进了对单乙二醇生产的选择性和对较高级的甘醇同系物形成的抑制-因此具有两种效果的益处。并且,这一点在具有最终产物的同时保持,最终产物在乙二醇中更加浓缩,因此在蒸发部分中需要的能量要少得多。例如,在本发明中,反应器部分的最终产物,即合并的反应器产物88,可以具有10:1或甚至更低的水:乙二醇的比率,而相比之下,在常规设施中,输送至蒸发器部分的乙二醇反应器流出物具有约25:1的水:环氧乙烷的摩尔比,因此将过量水蒸发所需的热负荷量减少至少60%。本发明的一个显著优点是可以根据催化剂和能量使用的需要和要求、环氧乙烷可用性和产物收率要求调节和优化水:环氧乙烷的摩尔比。已经说明了本发明的反应器部分的总体操作方案和益处,现在将继续参考图3更详细地描述本发明的方法。如上所述,流57和36合并形成流向第一反应器的进料流37,并且因此,通过在交换器35中控制对水流36的加热(这将进而控制流37的入口温度)来调节反应器38的催化剂床出口温度。连续串联的反应器40、46和52中的催化剂床出口温度各自通过使用分别用于冷却每个反应器的进料流的交换器39、43和49来调节。调节温度和压强是本发明的方法的重要部分。本发明中的水合反应以液相方法进行。在本发明中,“液相法”是指供应给反应器的进料流、反应器产物或反应器流出物流和反应器内的反应物保持在液相状态。因此,每个反应器中的温度和压强使得当流供应到每个反应器并进入每个反应器时,它们保持在液相状态。因此调节每个反应器中的温度和压强以使反应器的内容物保持在液相状态。温度的选择在很大程度上取决于上述段落的考虑因素。因此,在本发明中给定下面的温度范围,反应器内容物为液体形式,压强将为介于约0.5mpa至约1.5mpa之间,优选0.8mpa至1.2mpa之间。在选择温度时,必须平衡两个相互竞争的要求。首先,本发明中入口的温度必须足够高以驱动环氧乙烷的水合反应,以使环氧乙烷向单乙二醇的转化完全或接近完全。具体地,“完全”(“complete”)、“接近完全”或“完全”(“completion”)是指环氧乙烷转化为单乙二醇的转化百分比应为至少97%,优选至少约99%。因此,反应器38、40、46和52的入口温度必须在约50℃至约90℃的范围内,优选在约70℃至约85℃的范围内。然而,反应器中(以及因此在反应器出口处)的温度不应太高,因为如上所述,反应器内部的温度显著升高导致离子交换树脂的溶胀和降解。反应器中的高温是放热水合反应的结果,因此为了缓和任何温度升高,必须除去这种反应热。在这些应用中,绝热反应器具有相当少的方法复杂性,但缺点是它们不能除去反应热。因此,在本发明中已经发现,尽管如此,温度升高仍可以通过每个反应器中作为散热器的过量水来至少部分地缓和。因此,尽管本发明的一个目的是减少用于生产乙二醇的水量,但是至少需要最少量的水来缓和由放热反应引起的反应器中的温度升高。如上所述,在本发明中这两个目标通过以下方式满足:将含水环氧乙烷进料流40分成四个相等的部分57、61、64和67并且将它们共同进给(也可以使用术语“分级进给”)以形成流向反应器38、40、46、52的进料流。利用该发明配置,在每个反应器内部实现高水:环氧乙烷比率,从而缓和放热,使得在每个反应器的出口处温度介于约85℃至约120℃之间,优选介于约100℃至约110℃之间。尽管本发明在缓和反应器中的温度升高方面取得了成功,但可能仍然需要进一步降低反应器流出物的温度直至达到上述可接受的入口温度范围。为此,流70、73和76分别通过热交换器39、43和49。这提供了热集成的机会,因为来自反应部分外部的流动流(未图示)可以通过与来自一个或多个串联反应器的反应器流出物的间接热交换而在任何热交换器39、43和49中加热。如上所述,在较高温度下,环氧乙烷几乎完全转化为单乙二醇-在较低的入口温度下,并非所有的乙二醇都会反应,并且在反应器流出物中可检测量的环氧乙烷将出现并保持未反应(“突破”)。在某些情况下,可能希望降低入口温度以降低环氧乙烷转化率,从而使得更多的环氧乙烷能突破-例如当希望延长离子交换树脂催化剂的寿命时或在其他情况下,如下面所描述的。除了上述参数和操作条件之外,在操作该方法时可以考虑的其他因素包括但不限于:反应器和方法配置、转化率和离子交换树脂催化剂的选择性、以及反应物流速(lhsv)。这种独特的反应器配置使得在选择最适合设施操作目标的关于催化剂和能量使用、环氧乙烷可用性和产物产率要求的反应器操作参数方面能有灵活性。此外,与由于常规的非催化热水解需要更加稀释的环氧乙烷水溶液和高温而导致需要更多步骤的传统商业设施相比,本发明的制备进给的高浓度环氧乙烷液体进料流40显著减少了环氧化方法部分所需的步骤数。考虑到上述考虑因素,在合适的温度下,进料流37进入反应器38,在反应器38中,环氧乙烷在离子交换树脂催化剂或催化剂床的存在下与水反应形成单乙二醇。在本发明的绝热反应器中,例如反应器38中,将环氧乙烷与热稀释水混合以在催化剂存在下达到适当的温度,导致对单乙二醇具有高选择性的环氧乙烷的水合,使得至少约95%,优选至少约98%的环氧乙烷转化成单乙二醇,只有很小的百分比被转化成高级甘醇如二甘醇或三甘醇。因此,例如对于反应器38,就摩尔数而言,反应器流出物70中单乙二醇的摩尔数将近似于进料流38中环氧乙烷的摩尔数。除了单乙二醇产物外,本发明中的反应器的每种流出物还含有水(用于下一反应器的过量未反应的水)、单乙二醇和痕量的其他组分,其他组分包括环氧乙烷和高级甘醇同系物。尽管在反应器38的整个长度上存在压强下降,但是反应器38和40之间存在足以使第一反应器流出物70通过热交换器39的压强差,在热交换器39将其冷却,然后将第一反应器流出物与流61合并以形成进入反应器40的第二进料流81,如同在反应器38中,环氧乙烷与水以高选择性和转化率反应,使得基本上所有环氧乙烷与化学计量的水反应,以按上述转化百分比产生单乙二醇。类似于第一反应器流出物,第二反应器流出物含有未反应的水、单乙二醇和上述的痕量组分,借助于压差行进通过热交换器43。基本上第一反应器流出物70和第二反应器流出物73之间的唯一区别是后者含有更多的单乙二醇,因为它含有在反应器38和40中产生的单乙二醇的量。反应器46和52和它们的相关流以与反应器38和40的操作方式相同的方式操作,其中第二反应器流出物73在热交换器43中冷却,并与相等部分64的含水浓缩环氧乙烷进料流40合并形成第三进料流75,第三进料流75在第三反应器46中在第三反应器树脂催化剂床存在下反应而形成第三反应器流出物76。第三反应器流出物76在热交换器49中冷却并与相等部分67合并以形成第四进料流77,第四进料流77在第四反应器52中在第四离子交换树脂催化剂床存在下反应,以形成第四反应器流出物79,每个流出物如上所述逐渐变得更富含单乙二醇。除了先前描述的包围四个向下流动式的固定床串联反应器的反应器序列22之外,反应器部分25还包括传统的管式反应器26,第四反应器流出物79可在热交换器55中加热后供应到该管式反应器26中。在本发明中,该管式反应器26被配置和操作以提供迄今未知水平的对催化水合方法方案的通用性和可操作性,现在将对其更详细地描述。首先,除了提高环氧乙烷和水转化成meg的转化率及其对meg的选择性之外,本发明还可以操作和配置以通过使得方法操作者能制造二甘醇和三甘醇来提高水合方法的多功能性和适应性。这可以通过物理地绕过催化反应器38、40、46、52中的一个或多个(该旁路本身未示出)并且因此从进料流的循环中除去一个或多个催化反应器来实现。在绕过上述催化剂反应器中的一个或多个之后“突破”的保持未反应的环氧乙烷被输送至非催化管式反应器26,以将环氧乙烷和水热转化为乙二醇。由于管式反应器26是非催化的并且完全依赖于热转化,因此第四反应器流出物79在热交换器55中被预热以达到有效温度范围内,从而将环氧乙烷非催化水合成单乙二醇-优选管式反应器26的入口温度为约130℃至约160℃。当然,这种非催化热转化对meg的选择性显著降低,其中,还相对于meg产生大量高级的甘醇如deg和teg,从而通过增加在管式反应器中转化为乙二醇的环氧乙烷的量,与反应部分25中仅使用催化反应器时将产生的meg和deg和teg相比,合并反应器产物88含有较少的meg和较多的deg和teg。这为操作者提供了用于乙二醇生产的通用且灵活的方法,因为操作者可以调节单乙二醇、二甘醇和三甘醇产物的产量以满足需求。在极限情况下,绕过所有四个串联反应器,使得仅留下最终的非催化管式反应器26并且在该反应器中将发生所有水合反应-从而将催化方法转化为非催化方法。替代地,与使用非催化eo水解的设施中的相同生产相比,催化序列22增加了灵活性以减少设施中的有用物质(utility)消耗,同时仍保持固定的单乙二醇生产。如果在反应器序列22的催化反应器38和40(绕过反应器46和42)中进行乙二醇的生产的约一半,并且在管式热反应器26中进行平衡,则获得的设施产物分布将是类似于传统的非催化热水解方法,但具有使设施以低约20%摩尔比的水:环氧乙烷运行的优点,导致乙二醇蒸发中的有用物质的显著节省。替代地,不是物理地绕过反应器,而是通过将一个或多个串联反应器的入口温度降低到与所期望的一样低来实现“温度旁路”,以减少环氧乙烷和水之间的反应并减少在反应器中制备的单乙二醇的量,这意味着在催化反应器的出口中看到更多未反应的环氧乙烷“突破”。然后如上所述将该环氧乙烷在管式反应器中转化为乙二醇。管式反应器还可以以“精加工”的角色起作用。即使当反应器序列22在按所设计的正常催化效率下操作以使环氧乙烷转化为乙二醇的转化率最大化时,但是仍然有少量未反应的环氧乙烷通过序列22中的所有四个反应器保持未反应,并且该环氧乙烷将在第四反应器流出物79中“突破”。在本发明中,优选操作序列26,使得在第四反应器流出物79中的环氧乙烷的浓度将不大于约1mol%。然后可将该剩余的环氧乙烷在管式反应器26中转化为单乙二醇。最后,在催化反应器中的一个或多个发生故障或由于耗尽、树脂床或喷嘴堵塞或者在离子交换树脂床操作中常见的一些其他困难而必须以其他方式从使用中移除的情况下,管式反应器26也可用作备用反应器。当发生这种情况时,必须停止使用有故障的催化反应器。这意味着在反应部分22中存在过量的未转化的环氧乙烷,这是不希望有的。这种过量的环氧乙烷突破可以在管式反应器26中转化为乙二醇。因此,在本发明中存在管式反应器26为方法操作者提供了相当大的灵活性和可操作性。与不能更换或修复离子交换树脂催化剂或反应器的现有技术不同的是固体/非均相催化体系的严重缺点,在本发明中,反应器可以离线进行这种修复而不中断操作。虽然在图2和3的反应器部分中示出了四个反应器和三个热交换器,但这仅用于说明的目的。根据具体需要和要求,可以使用更多或更少的反应器。增加反应器的数量具有减少每个反应器的放热的优点-从而减缓了离子交换树脂催化剂在每个反应器内经历的温度引起的不可逆溶胀的量。另外,增加反应器的数量也会增加环氧乙烷的稀释度,并且因为环氧乙烷降解大多数离子交换树脂,所以预期增加的稀释会延长树脂的寿命。此外,降低了总催化剂体积要求。更多反应器的主要缺点是成本和复杂性,包括反应器、泵、交换器和其他设备的额外资金成本以及更大体积的离子交换树脂催化剂以及额外的管道、仪器的操作成本和操作复杂性。然后可以通过多效蒸发器或真空蒸馏或本领域技术人员已知的任何其他合适的方法回收合并的反应器产物88中的亚烷基二醇。可以将回收技术组合用于回收中的不同阶段。例如,多效蒸发器可用于从反应器流出物中除去水,而真空蒸馏可用于甘醇干燥。实际上,本发明的益处之一是,在设施的后甘醇反应部分中,因为已经减少了蒸发过量水所需的热负荷的量,所以复杂性可以降低,如上所述并且如在下面的示例中详细描述。因此,尽管现有技术的设施通常包含具有若干效果或阶段的蒸发系统(参见例如epno.1121646b1),但本发明需要较少数量的阶段。如上所述,本发明可涉及一种或多种离子交换树脂催化剂。离子交换树脂具有聚合物基质,其在表面上含有由离子官能团填充的离子交换位点。离子交换树脂通常在阳离子交换树脂或阴离子交换树脂之间进行区分,但也可以使用其他类型的离子交换树脂。有关阳离子交换树脂和阴离子交换树脂的更全面的描述,参见dedardel,f.andarden,t.v.ionexchangersinullman’sencyclopediaofindustrialchemistry(2005)。用于离子交换树脂催化剂的合适的聚合物基质包括聚苯乙烯基质、聚丙烯酸基质、聚烷基胺树脂以及其他聚合物材料。优选地,聚合物基质与二乙烯基苯(divenylbenzene)交联至足以提高操作容量的程度,同时还不将离子交换材料的密度增加到离子交换材料变得太物理硬化和对化学处理太耐化学性的程度。优选地,基质是苯乙烯,二乙烯基苯(divenylbenzene)共聚物。固定到上述聚合物基质上的位点是离子固定基团,其确定树脂是否起阳离子或阴离子离子交换树脂的作用。在溶液中,固定基团的正电荷或负电荷由相反电荷的离子补偿,这些离子在本文中称为官能团。强酸性阳离子离子交换树脂通常包括作为固定基团的磺酸基团,其依次与苯乙烯-二乙烯基苯聚合物基质连接。强酸性磺酸阳离子交换树脂的示例包括amberliteir120、dowexhcr、lewatits100和amberlyst15等。阳离子离子交换树脂还可包括树脂材料,其是通过丙烯酸或甲基丙烯酸和二乙烯基苯的加成聚合获得的共聚物,如美国专利3,957,698中所公开的。用于阳离子交换树脂的其他合适的聚合物材料包括当聚乙烯吡啶树脂与二乙烯基苯交联时形成的树脂基质。此类材料可以以reillexhpq商标获得。磺化酚醛聚合物树脂也是合适的阳离子离子交换树脂。弱酸性阳离子离子交换树脂通常包含羧基作为固定基团。弱酸性阳离子交换树脂的示例包括amberliteirc86、dowexmac-3、lewatitcnp等。如本文中所使用的,在本发明中,强碱性阴离子交换树脂含有季铵固定基团。这些进一步分为i型和ii型,i型通过氯甲基化后三甲胺与苯乙烯-二乙烯基苯共聚物的反应制备,ii型通过苯乙烯-二乙烯基苯共聚物与二甲基乙醇胺反应得到。这种i型树脂的合适示例包括可购自lanxess的lewatitmp500,和amberlyst26和amberliteira402,以及可购自dow的ira410。强碱性ii型树脂的合适示例包括例如purolitea510s(purolitecorporation)。弱碱性阴离子交换树脂通常包括聚丙烯酸树脂,该聚丙烯酸树脂通过与多官能胺反应而具有固定基团,以产生阴离子交换树脂,例如叔铵弱碱性amberliteira67和amberlyst21树脂(购自dow)。应特别注意的是,然后可以用氯甲烷或硫酸二甲酯进一步处理该离子交换树脂,得到季胺强碱性i型树脂amberliteira458树脂(dow)。弱碱性阴离子交换树脂还可包括作为固定基团的游离碱基团,例如amberliteira-67树脂(dow)。在本发明的一具体实施方案中,强碱性阴离子交换树脂在季铵固定基团和聚合物基质/材料的苯基团之间含有具有3-7个连接原子,优选3-5个碳原子的“连接”基团。在强碱基树脂中的这种连接链的示例在美国专利no.5,945,568中说明,并以diaiontsa1200(mitsubishichemical)的名称生产。如no.5,945,568专利中所述,将季铵固定基与聚合物基质的苯基连接的连接基团没有特别限制,只要其足够长即可。连接基团的合适示例是亚烷基或亚烷基氧亚烷基。用于本发明的优选示例是阴离子交换树脂,其包含作为基底的乙烯基芳族化合物的聚合物,并且其结构使得季铵基团通过链长至少为3的连接基团的芳族基团键合。不受理论的限制,相信在本发明中,该连接基团防止苯环对铵环固定基团的亲核攻击,否则其会导致离子交换树脂催化剂的加速失活和降解以及稳定性降低和催化剂寿命缩短。多种离子适合用作本发明的阴离子树脂中的官能团,并且可以选自碳酸氢盐、碳酸盐、氢氧化物、亚硫酸氢盐、羧酸盐、柠檬酸盐和金属盐,以及钼酸根阴离子。这些官能团可以与上面确定的任何固定基团和树脂材料一起使用。在本发明中,优选离子交换树脂催化剂来自i型强碱性阴离子交换树脂,更优选具有碳酸氢盐或单柠檬酸盐官能团的i型强碱性阴离子交换树脂,并且最优选具有碳酸氢盐或单柠檬酸盐官能团与连接基团的i型强碱性阴离子交换树脂。除了上面指定的离子交换材料之外,可以使用任何其他合适的阳离子或阴离子交换树脂,例如上述dedardel,f.andarden,t.v.2008,ionexchanger,inullman’sencyclopediaofindustrialchemistry中所述的阳离子和阴离子交换树脂。除了上述离子交换材料之外,还可以使用均相和非均相催化剂的合并-例如上面吸附有可溶性金属催化剂的固体催化剂。在这种情况下,首先制备上面吸附有金属催化剂的固体催化剂。然后,在上述液相水合反应期间,金属催化剂从固体催化剂中解吸并可有效地催化反应。金属催化剂然后必须与反应产物分离,并且可以重新用于再吸附到固体催化剂上。在这种情况下,固体催化剂可以是离子交换树脂。以上段落涉及选择适用于本发明的催化反应器的离子交换树脂催化剂。关于含离子交换树脂的催化反应器的操作,在图2和3中,进料流沿从反应器顶部向下穿过每个反应器的方向行进。这被称为“向下流动”模式,但在本发明中,反应器可以以“向下流动”或“向上流动”模式操作。下流模式方法具有的优点是增加了反应器内催化剂的密度或体积,从而减小了反应器本身的尺寸和成本。向下流动操作还使水和环氧乙烷成为乙二醇的非催化反应最小化。然而,在向下流动操作过程中,催化剂选择性可能受到局部不均匀性和可能产生并陷入催化剂床中的杂质以及受到催化剂床中产生的“沟道作用”(“channeling”)的影响。可以推测,在向上流动操作中,进料流沿着从反应器底部向上的方向行进。在pct公开号wo2008/150338a1中,报道了在向上流动操作模式下离子交换树脂的稳定性增强和使用寿命延长。在本发明中,液相反应物和固体催化剂向上流动操作可以在向上流动操作期间提供适量的床流化,向上流动操作增加了相邻树脂颗粒之间的空隙率,从而允许减少不均匀性和污染,否则在向下流动操作期间嵌入并捕获在催化剂床中并降低催化剂性能。美国专利no.6,160,187先前已经贬低了催化反应器的向上流动操作,因为所得到的床流化据称由于轴向混合而导致更大的催化剂磨损和降低的单乙二醇选择性。然而,在本发明中已经发现,在本发明的液相方法中,这些批评是不现实的,因为它们更可能发生在气体/固体催化剂体系中,其中存在更高的速度,(与液体反应物和固体催化剂之间的密度差异相比,在气体反应物和固体催化剂之间有)更大的密度差异,因此与我们的发明的本发明液体/固体催化剂体系中的床溶胀相比有大得多的床溶胀。因此,向上流动操作仍然是本发明中的合适操作模式。银基环氧化催化剂银基环氧化催化剂包含载体和至少催化有效量的银或含银化合物;还任选存在的是促进量的铼或含铼化合物;还任选存在的是促进量的一种或多种碱金属或含碱金属的化合物。在本发明中使用的载体可以选自大量的可以是多孔的并且可以提供优选孔结构的固体耐火载体。氧化铝是公知的用作烯烃的环氧化的催化剂载体,并且是优选的载体。不论使用的载体的性质如何,载体都通常成形为具有适合在固定床环氧化反应器中应用的尺寸的颗粒、厚块(chunk)、片状、丸状、环状、球体状、车轮状、横向分割的空心圆柱体等等的形状。载体颗粒的等效直径优选在从约3mm至约12mm的范围内,并且更优选在约5mm至约10mm的范围内。(等效直径为与被采用的载体颗粒具有相同的外表面(即,忽略颗粒的孔隙内的表面)比体积的比率的球体的直径。)合适的载体可从saint-gobainnorproco.,sudchemieag,noritakeco.,ceramtecag和industriebitossis.p.a.获得,不限于其中包含的具体组合物和制剂,关于载体组合物和制备载体的方法的进一步信息可以在美国专利公开号2007/0037991中找到。为了制备用于烯烃氧化成烯烃氧化物的催化剂,然后在具有上述特性的载体的表面上提供催化有效量的银。在一实施方案中,催化有效量的银为10重量%至45重量%。该催化剂通过用溶解在足以使银前体化合物沉积到载体上的合适的溶剂中的银化合物、络合物或盐浸渍载体来制备。优选地,使用含水的银溶液。促进量的铼组分也可以在银的沉积之前、同时、或之后沉积在载体上,所述铼组分可以是含铼化合物或含铼络合物。基于包括载体的总催化剂的重量计,以铼金属表示的铼促进剂可以存在的量为约0.001重量%至约1重量%,优选约0.005重量%至约0.5重量%,更优选为约0.01重量%至约0.1重量%。在银和铼的沉积之前、同时、或之后,在载体上还可沉积的其他组分是促进量的碱金属或两种或两种以上碱金属的混合物,以及任选的促进量的iia族碱土金属组分或两种或两种以上iia族碱土金属组分的混合物,和/或过渡金属组分或两种或两种以上过渡金属组分的混合物,所有这些都可以是在适当的溶剂中溶解的金属离子、金属化合物、金属络合物和/或金属盐的形式。可以用各种催化剂促进剂在同一时间或在不同的步骤中浸渍该载体。相比于银和载体以及无促进剂或仅一种促进剂的相同的组合,本发明的载体、银、碱金属促进剂、铼组分和任选的额外促进剂的特定组合将提供一种或多种改进的催化性质。如本文所采用的术语,催化剂的某种组分的“促进量”是指在与不含所述组分的催化剂相比时,该组分的量可有效地改进所述催化剂的催化性质。所采用的确切的浓度当然将尤其取决于所期望的银含量、载体的性质、液体的粘度、和用于输送促进剂到浸渍溶液的特定化合物的溶解度。催化性质的示例尤其包括可操作性(抗失控性)、选择性、活性、转化率、稳定性和产率,以及其他催化性能。本领域技术人员所理解的是,一种或多种单独的催化性质可以通过“促进量”来增强,而其他催化性质可以或可以不被增强甚至可以被减弱。适当的碱金属促进剂可以选自锂、钠、钾、铷、铯或它们的组合,优选的是铯,特别优选的是铯与其他碱金属的组合。沉积或存在于载体上的碱金属的量是促进量。优选地,该量的范围按总催化剂的重量计,按该金属计量为约10ppm至约3000ppm,更优选为约15ppm至约2000ppm,甚至更优选为约20ppm至约1500ppm,并且特别优选为约50ppm至约1000ppm。适当的碱土金属促进剂包括选自元素周期表的iia族的元素,其可以是铍、镁、钙、锶和钡或它们的组合。合适的过渡金属促进剂可包括元素周期表第iva、va、via、viia和viiia族的元素,及其组合。沉积在载体上的碱土金属促进剂和/或过渡金属促进剂的量是促进量。过渡金属促进剂通常可存在的量为每克约0.1微摩尔至每克约10微摩尔,优选为每克约0.2微摩尔至每克约5微摩尔。用于浸渍载体的银溶液也可包括如本领域中公知的任选的溶剂或络合剂/增溶剂。多种溶剂或络合剂/增溶剂可用于在浸渍介质中溶解银至所需的浓度。有用的络合剂/增溶剂包括胺、氨、草酸、乳酸和它们的组合物。胺包括具有1至5个碳原子的亚烷基二胺。在一优选的实施方案中,溶液包括草酸银和乙二胺的水溶液。络合剂/增溶剂存在于浸渍溶液的量可以为每摩尔银约0.1至约5.0摩尔,优选每摩尔银约0.2至约4.0摩尔,并且更优选每摩尔银从约0.3至约3.0摩尔。当使用溶剂时,所述溶剂可以是有机溶剂或水,并且可以是极性的或基本上或完全非极性的。通常,溶剂应具有足够的溶剂化能力以溶解溶液组分。同时,优选的是溶剂被选择成避免对溶剂化促进剂具有不恰当的影响或与溶剂化促进剂有相互作用。具有每分子1至约8个碳原子的有机基溶剂是优选的。也可以使用数种有机溶剂的混合物或有机溶剂与水的混合物,条件是这些混合的溶剂如本文所希望的发挥作用。在浸渍溶液中的银的浓度典型地是在约0.1重量%至通过所使用的特定的溶剂/增溶剂组合物所提供的最大溶解度的范围内。采用含有0.5重量%至约45重量%的银的溶液通常是非常合适的,优选含有从5重量%至35重量%的浓度的银的溶液。使用任何常规的方法来实现所选择的载体的浸渍;例如,过量溶液浸渍、初湿含浸法、喷涂法等等。典型地,载体材料被放置成接触含银溶液,直至载体吸收足够量的所述溶液为止。优选用于浸渍多孔载体的含银溶液的量不超过填充载体孔隙所需的量。部分取决于在所述溶液中的银成分的浓度,可采用单次浸渍或一系列浸渍,其中对中间产物进行干燥或不对中间产物进行干燥。浸渍操作方法描述于例如美国专利no.4,761,394、no.4,766,105、no.4,908,343、no.5,057,481、no.5,187,140、no.5,102,848、no.5,011,807、no.5,099,041和no.5,407,888中。还可采用用于对各种促进剂预沉积、共沉积和后-沉积(post-deposition)的已知现有操作方法。在用含银化合物(即,银前体)、铼组分、碱金属组分和任选的其他促进剂浸渍所述载体之后,将浸渍的载体煅烧足够的时间以将含银化合物转化成活性银物质,并从经浸渍的载体除去挥发性组分以产生催化剂前体。可通过加热经浸渍的载体实施所述煅烧,其是优选以逐渐增大的速率、在约0.05至约3.5mpa的范围内的压强下加热至约200℃至约600℃的范围内的温度来实施。一般而言,温度越高,所需的加热持续时间越短。本领域中已经提出了宽范围的加热持续时间;例如,美国专利us3,563,914公开了加热持续时间少于300秒;以及us3,702,259公开了在100℃至375℃的温度下加热2至8小时,通常持续时间为约0.5至约8小时。但是,唯一重要的是,加热时间与温度相关联,使得基本上所有包含的银被转化成活性银物质。连续或分步加热也可以用于此目的。在煅烧期间,经浸渍的载体可以暴露于包含惰性气体或惰性气体与按体积计约10ppm至21%的含氧氧化组分的混合物的气氛。对于本发明的目的,惰性气体被定义为在选择用于煅烧的条件下基本上不与催化剂或催化剂前体反应的气体。在上述的美国专利公开no.2007/0037991中可找到关于催化剂制备的进一步信息。仅为了说明的目的,以下是在目前的商业环氧乙烷反应器单元中通常使用的条件:气体时空速率(ghsv)1500-10000h-1,反应器入口压强1mpa至3mpa,冷却剂温度180-315℃,氧转化水平10-60%,以及eo生产速率(工作速率)100-350kgeo/m3催化剂/小时,环氧乙烷浓度的变化,δeo,为约1.5%至约4.5%。启动完成后和在正常操作期间在反应器入口的进料组合物通常包含(按体积百分比计)1-40%的乙烯,3-12%的o2;0.2%至10%,优选0.2%至6%,更优选为0.2%至5%的co2;0-5%的乙烷;在本文中描述的一定量的一种或多种氯化物缓和剂;以及进料的余量包括氩气、甲烷、氮气或它们的混合物。实施例现在将参考以下非限制性实施例更详细地描述本发明。实施例1(现有技术)使用单个非催化管式/管道式反应器模拟根据现有技术制备乙二醇的方法。在该方法中,将进料流进给到反应器中,从反应器中取出反应器流出物,下表给出以千克/小时为单位的值:原料流反应器流出物温度150--190环氧乙烷1000--0水10191--9798meg45--1315重甘醇0--124eo转化率(%)--100%--weor(mol)25----如上所使用的,“weor”是水比环氧乙烷的摩尔比。该现有技术方法以良好的转化率生产乙二醇,但使用大量过量的水-该过量的水然后必须从乙二醇产物中除去,使得该方法与根据本发明的方法相比,能量明显更加密集,如下面将讨论的。实施例2(本发明)根据图4模拟根据本发明的制备乙二醇的方法。该实施方案与上述图2中的反应序列25类似地配置,其中常规管式热反应器26相当于管式反应器“dd”。如图4所示,在模拟中,存在四个向下流动式的固定床系列布置的催化反应器,其具有作为加热装置的三个级间在线热交换器。在该模拟中,反应器填充有一种或多种如上所述的合适的离子交换树脂。该方法根据本发明操作,使得流、入口和出口点和反应器的温度和组成如下表i所示,其中表中流的含量以千克每小时为单位给出。从上表中可以看出,水和环氧乙烷以10:1的摩尔比(流a和b的组合)提供给该方法-这比现有技术方法低得多,例如如上面的实施例1中所示。然而,通过将输入流分成进给到本发明所规定的多个反应器的多个流,在每个反应器中获得了远远更高的水比环氧乙烷的比率-如上所示,进入反应器中的流中水比环氧乙烷的摩尔比在每种情况下约为32:1。这种高weor不仅提高了环氧乙烷对meg的选择性和环氧乙烷转化为meg的转化率(如上图所示,在此模拟中转化为meg的转化率为99%),但它也有助于缓和每个反应器中的温度升高,从而减少级间热交换器中所需的冷却量和降低可能潜在地损坏离子交换树脂催化剂的高温。此外,如上所述,与现有技术相比,在根据本发明制备的方法中使用较低的weor导致相当大的能量节省。在根据本发明的方法的模拟(实施例2)和对比现有技术方法(实施例1)的模拟中,最终的蒸发负荷是:整体weor蒸发负荷(tph)本发明(实施例2)10:1196现有技术(实施例1)25:1523因此,现有技术方法的蒸发器负荷是本发明的2.5倍。本领域技术人员应理解,可以对上述液相方法的实施方案进行改变而不背离其广泛的发明构思。因此,应理解,本发明不限于所公开的特定实施方案或配置或组合物,而是意在涵盖由所附权利要求限定的本发明的精神和范围内的修改。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1