酰胺的制备方法与流程
[0001]
本发明涉及酰胺的制备方法。
[0002]
本发明主张基于2018年6月15日在日本申请的特愿2018-114577的优先权,在此引用其内容。
背景技术:
[0003]
在肽合成中,将氨基酸的羧基活化,使其与氨基酸的氨基发生偶联反应而形成酰胺键,通过重复该反应来使氨基酸依次延伸。作为使羧基活化的方法,已知有几种方法。存在使用活化度弱的缩合剂对异构化及副产物的生成进行抑制的同时合成肽的方法;也存在使用活化剂在短时间内合成肽的方法。
[0004]
作为使用高活性的活化剂使所述羧基活化的方法,有酰氯法、酸酐法。这些酰氯法、酸酐法与对活化度弱的缩合剂加以使用的活化法相比,由于活化剂的结构简单,因此具有单价便宜并且来自活化剂的副产物的生成少等优点。
[0005]
酸酐法分为对称酸酐法和混合酸酐法。
[0006]
例如,在非专利文献1~非专利文献2中,公开了使用对称酸酐作为羧酸活性物质的酰胺合成法。
[0007]
非专利文献1~非专利文献2中公开的对称酸酐法可称为具备以下步骤的方法:第1步骤,通过羧酸之间的缩合反应生成对称酸酐;第2步骤,进行上述对称酸酐与胺的偶联反应。
[0008]
此外,例如在非专利文献3中,公开了使用混合酸酐作为羧酸活性物质的酰胺合成法。
[0009]
在非专利文献3中,记载了将羧酸和氯甲酸异丙酯在第1微型混合器中混合,在短时间内合成混合酸酐,接着,为了不使合成的混合酸酐外消旋化,立即将含有混合酸酐的溶液同胺以及催化剂(碱)在第2微型混合器中混合,进行酰胺化。
[0010]
非专利文献3中公开的混合酸酐法可称为具备以下步骤的方法:第1步骤,使羧酸与氯甲酸酯反应得到混合酸酐;第2步骤,向上述混合酸酐中添加碱得到酰基吡啶鎓物质;第3步骤,将所述酰基吡啶鎓物质与胺进行偶联反应得到酰胺。
[0011]
现有技术文献
[0012]
非专利文献
[0013]
非专利文献1:“efficient amide bond formation through a rapid and strongactivation of carboxylic acids in a microflow reactor”,fuse,s.mifune,y.takahashi,t.,angew chem.int.ed.53,851-855(2014)。
[0014]
非专利文献2:“total synthesis of feglymycin based on a linear/convergent hybrid approach using micro-flow amide bond formation”,fuse,s.mifune,y.nakamura,h.tanaka,h.nat.commun.7,13491(2016)。
[0015]
非专利文献3:小竹佑磨、中村浩之、布施新一郎,
“マイクロフロー
法
を
駆使
する
n-
メチル
化
ペプチドの
効率的合成”,2017年3月16日,日本化学会第97春季年会、3f4-14。
技术实现要素:
[0016]
本发明所解决的技术问题
[0017]
但是,在对称酸酐法中,由于对称酸酐与胺的反应性低,所以存在与亲核性低的胺的偶联反应花费较多时间或者反应无法进行这样的问题。
[0018]
另外,在混合酸酐法中,存在由于酰基吡啶鎓物质与酰基吡啶鎓物质的平衡阴离子的反应,生成不期望的化合物(即,酯)这样的问题。
[0019]
本发明是为了解决上述问题而完成的,其目的在于提供下述的酰胺制备方法:在将羧基活化使其与氨基发生偶联反应而形成酰胺键的反应中,反应效率良好且难以发生副反应。
[0020]
解决技术问题的技术方案
[0021]
也就是说,本发明具有以下方式。
[0022]
(1)一种酰胺的制备方法,该方法包括在使羧酸之间发生脱水缩合后,再与碱进行反应,然后与胺进行反应。
[0023]
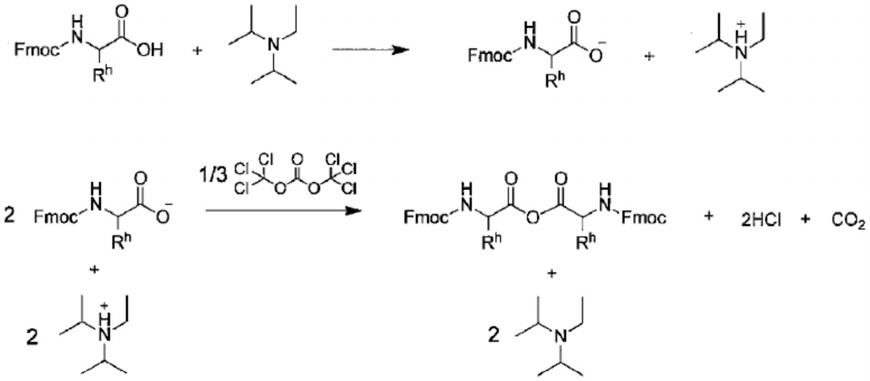
(2)一种酰胺的制备方法,该方法包括使将第一羧酸和第二羧酸混合得到的混合物进行反应,再将该反应的产物同碱和胺进行混合。
[0024]
(3)根据上述(1)或(2)所述的酰胺的制备方法,使光气或者在反应体系内分解后生成光气的光气等价物反应,使所述羧酸之间脱水缩合。
[0025]
(4)根据上述(1)~(3)中任一项所述的酰胺的制备方法,使相同种类的羧酸之间脱水缩合。
[0026]
(5)根据上述(1)~(4)中任一项所述的酰胺的制备方法,所述羧酸是氨基酸或氨基酸衍生物。
[0027]
(6)根据上述(1)~(5)中任一项所述的酰胺的制备方法,所述碱是选自于由吡啶、吡啶衍生物、咪唑、咪唑衍生物以及1,4-二氮杂双环[2,2,2]辛烷组成的组中的任意一种以上。
[0028]
(7)根据所述(1)~(6)中任一项所述的酰胺的制备方法,所述碱是选自于由4-吗啉基吡啶、n,n-二甲基-4-氨基吡啶、4-吡咯烷基吡啶、吡啶、4-甲氧基吡啶、咪唑、n-甲基咪唑以及1,4-二氮杂双环[2,2,2]辛烷组成的组中的任意一种以上。
[0029]
(8)根据上述(1)~(7)中任一项所述的酰胺的制备方法,所述胺是氨基酸或氨基酸衍生物。
[0030]
(9)根据上述(1)~(8)中任一项所述的酰胺的制备方法,所述胺的亲核性低于从构成蛋白质且作为遗传信息编码的20种氨基酸中除去缬氨酸和异亮氨酸的18种氨基酸的亲核性。
[0031]
(10)根据上述(8)或(9)所述的酰胺的制备方法,所述胺是缬氨酸、异亮氨酸、或n-烷基化的氨基酸或者上述物质的衍生物。
[0032]
(11)根据上述(1)~(10)中任一项所述的酰胺的制备方法,在流通系反应装置中进行与所述胺的反应。
[0033]
(12)根据上述(11)所述的酰胺的制备方法,在使所述羧酸之间脱水缩合后,在流
通系反应装置中还进行与所述碱的反应。
[0034]
发明效果
[0035]
根据本发明,可以提供反应效率良好、难以发生副反应的酰胺制备方法。
附图说明
[0036]
图1是示出了流通系反应装置1的概略构成的模式图。
具体实施方式
[0037]
以下,对本发明的酰胺制备方法的实施方式加以说明。
[0038]
<酰胺的制备方法>
[0039]
实施方式的酰胺制备方法包括使羧酸之间脱水缩合后,再与碱进行反应,然后与胺进行反应。
[0040]
实施方式的酰胺制备方法也可以包括使将第一羧酸和第二羧酸混合得到的混合物进行反应,再将该反应的产物同碱和胺混合。实施方式的酰胺制备方法也可以包括将第一羧酸、第二羧酸同光气或者在反应体系内分解后生成光气的光气等价物混合得到混合物,使该混合物反应得到的产物同碱和胺混合。在此,将第一羧酸和第二羧酸混合得到混合物并使该混合物反应得到的产物包括使第一羧酸和第二羧酸脱水缩合得到的酸酐。
[0041]
另外,所述碱可以是生成阳离子活性物质的物质,也可以是碱(但所述胺除外)。
[0042]
另外,这里所说的“混合”是指在反应体系中添加原料等物质的动作,在反应体系内将它们混合时,原料等也可变为与添加前不同的物质。
[0043]
该制备方法可以包括以下的工序1~3。
[0044]
工序1:使羧酸之间脱水缩合得到酸酐的工序。
[0045]
工序2:使在上述工序1中得到的上述酸酐与碱反应,得到阳离子活性物质的工序。
[0046]
工序3:使在上述工序2中得到的上述阳离子活性物质与胺反应,制备酰胺的工序。
[0047]
以下,对上述各工序进行说明。另外,本发明所述的酰胺制备方法的反应并不限定于下述各工序中举例示出的反应。
[0048]
<工序1>
[0049]
工序1是使羧酸之间脱水缩合得到酸酐的工序。
[0050]
羧酸只要是在分子末端具有羧基的物质即可,可以用下述通式(1)表示。
[0051]
[化1]
[0052][0053]
(式中,r
1
是氢原子或一价有机基团。)
[0054]
羧酸可以被去质子化而成为羧酸根离子,可用下述通式(1i)表示。
[0055]
[化2]
[0056]
[0057]
(式中,r
1
是氢原子或一价有机基团。)
[0058]
例如,可通过在反应体系内的n,n-二异丙基乙胺(diea)等亲核性低的碱的存在下,置入羧酸而实现羧酸的去质子化。
[0059]“在碱的存在下”是指例如在添加了碱的溶剂中。该碱的种类只要在反应体系内能够使羧酸去质子化,不受到特别限定。
[0060]
实施方式的酰胺制备方法的工序1使下述通式(1)表示的羧酸以及下述通式(1)
’
表示的羧酸之间脱水缩合,得到下述通式(2)表示的酸酐。所述酸酐例如可以通过使光气或者在反应体系内分解后生成光气的光气等价物同羧酸反应来得到。
[0061]
[化3]
[0062][0063]
(式中,r
1
和r
2
各自独立地为氢原子或一价有机基团。)
[0064]
光气等价物在反应体系内分解后生成光气,在合成反应上可以用作实质上与光气等同的物质。作为光气等价物,可以举出二光气、三光气等。
[0065]
所述脱水缩合可以使不同种类的羧酸之间脱水缩合,也可以使相同种类的羧酸之间脱水缩合。即,所述式(1)以及式(1)
’
中的r
1
与r
2
可以相同,也可以彼此不同。
[0066]
在r
1
与r
2
相同的情况下,通式(2)表示的酸酐是对称酸酐。在r
1
与r
2
相同的情况下,在后述的工序2中生成的阳离子活性物质的平衡阴离子与活化前的羧酸根离子相同。虽然存在平衡阴离子与阳离子活性物质自反应的情况,但是只要平衡阴离子与活化前的羧酸根离子相同,即使进行自反应,产物也与阳离子活性物质活化前的对称酸酐相同。
[0067]
因此,在r
1
与r
2
相同的情况下,具有如下的优点:在反应体系中得到的酰胺的种类均一,易于有计划地获得目标种类的反应产物。
[0068]
所述羧酸优选为氨基酸或氨基酸衍生物。此处的羧酸包括作为酸酐前体的羧酸。所述氨基酸优选为α-氨基酸。另外,由于构成生物体内的肽或蛋白质的氨基酸通常为l型,所以上述氨基酸优选为l型。所述α-氨基酸可以是下述通式(1-1)表示的化合物。
[0069]
[化4]
[0070][0071]
(式中,r
0
表示氨基酸的侧链。)
[0072]
所述氨基酸可以是在生物体内构成肽或蛋白质且作为遗传信息编码的20种氨基酸。作为这些氨基酸,可以举出丙氨酸、精氨酸、天冬酰胺、天冬氨酸、半胱氨酸、谷氨酰胺、谷氨酸、甘氨酸、组氨酸、异亮氨酸、亮氨酸、赖氨酸、甲硫氨酸、苯丙氨酸、脯氨酸、丝氨酸、苏氨酸、色氨酸、酪氨酸、缬氨酸。另外,上述氨基酸也可以是胱氨酸等未作为遗传信息编码的种类的氨基酸。
[0073]
例如,所述式(1-1)中的r
0
在上述氨基酸为丙氨酸的情况下为
“-
ch
3”,在甘氨酸的情况下为
“-
h”,在缬氨酸的情况下为
“-
ch(ch
3
)
2”,在异亮氨酸的情况下为
“-
ch(ch
3
)
ch
2
ch
3”。对于其它氨基酸也为同样情况。
[0074]
在所述式(1)以及(1)
’
为氨基酸的情况下,-r
1
以及-r
2
可以分别为-ch(r
0
)nh
2
。
[0075]
所述氨基酸也可以不是α-氨基酸。例如可以是β-丙氨酸等β-氨基酸。
[0076]
所述羧酸也可以是氨基酸衍生物。氨基酸衍生物可以是具有与氨基酸实质上等同的性质的化合物,可以是天然存在的天然型化合物,也可以是与天然型不同的具有修饰、加成、官能团的取代等改变等的化合物。
[0077]
作为具有与氨基酸实质上等同性质的情况的实例,可以举出能够被以氨基酸作为底物的酶获取或者能够同与氨基酸结合的分子进行结合的情况。
[0078]
作为氨基酸衍生物,可以举出在氨基酸中1个以上的氢原子或者基团被除此以外的基团(取代基)取代的物质。作为氨基酸衍生物的示例,可以举出官能团被保护基保护的保护氨基酸。保护基具有使反应性的官能团失活的作用。还可以对保护基进行去保护,使被保护的官能团恢复到保护之前的状态。在此,“官能团被保护”是指构成所述官能团的原子被保护基取代的情况。作为被保护基保护的部位,可以举出选自于由氨基、羧基以及侧链组成的组中的任意一种以上的部位。可以用保护基保护侧链中包含的官能团的1处或者2处以上。在该工序1中,为了防止羧基以外的反应性官能团的反应,优选氨基和/或侧链的官能团被保护。
[0079]
作为保护基的种类,没有特别的限定,可以根据被保护的官能团的种类适当地进行选择。例如,作为氨基的保护基,可以举出氨基甲酸酯类、磺酰胺类、酰基类、烷基类等保护基,且不限于此。
[0080]
作为氨基甲酸酯类的保护基,可以举出2-苄氧基羰基(有时简写为-z或者-cbz)、叔丁氧基羰基(有时简写为-boc)、烯丙氧基羰基(有时简写为-alloc)、2,2,2-三氯乙氧基羰基(有时简写为-troc)、2-(三甲基硅烷基)乙氧基羰基(有时简写为-teoc)、9-芴基甲氧羰基(有时简写为-fmoc)、对硝基苄氧基羰基(有时简写为-z(no
2
))、对联苯异丙氧基羰基(有时简写为-bpoc)等。
[0081]
作为磺酰胺类的保护基,可以举出对甲苯磺酰基(有时简写为-ts或者-tos)、2-硝基苯磺酰基(有时简写为-ns)、2,2,4,6,7-五甲基二氢苯并呋喃-5-磺酰基(有时简写为-pbf)、2,2,5,7,8-五甲基色满-6-磺酰基(有时简写为-pmc)、1,2-二甲基吲哚-3-磺酰基(有时简写为-mis)等。
[0082]
<工序2>
[0083]
工序2是使上述工序1中得到的上述酸酐与碱反应,得到阳离子活性物质的工序。
[0084]
实施方式的酰胺制备方法的工序2是使下述通式(2)表示的酸酐与b表示的碱反应,得到下述通式(4)表示的阳离子活性物质的工序。另外,在该反应中,作为阳离子活性物质的平衡阴离子,生成下述通式(5)表示的化合物。
[0085]
[化5]
[0086][0087]
(式中,r
1
和r
2
各自独立地为氢原子或一价有机基团)
[0088]
工序2中的所述碱用于与上述酸酐生成阳离子活性物质,优选亲核性高的物质,更
优选选自于由吡啶、吡啶衍生物、咪唑、咪唑衍生物以及1,4-二氮杂双环[2,2,2]辛烷组成的组中的任意一种以上。
[0089]
吡啶衍生物可以是吡啶的一个以上的氢原子被其它基团取代的物质,只要具有碱的性质即可,不受到特别限定,吡啶以及吡啶衍生物优选为下述通式(3-1)表示的化合物。
[0090]
[化6]
[0091][0092]
(式中,x
1
表示氢原子或者选自于由用下述式(a)~(c)表示的组中的任意基团。)
[0093]
[化7]
[0094][0095]
(式中,r
31
、r
32
、r
33
以及r
34
各自独立地表示烷基。r
33
和r
34
可以相互键合形成环,r
33
或r
34
中未直接键合的、上述烷基中的1个亚甲基可以被氧原子替换。)
[0096]
r
31
、r
32
、r
33
以及r
34
的所述烷基可以是直链状、支链状和环状中的任一种。在环状的情况下,上述烷基可以是单环状或者多环状的任一种。所述烷基的碳原子数可以为1~20,也可以为1~15,还可以为1~10。
[0097]
作为直链状或者支链状的所述烷基,可以举例示出甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、异戊基、新戊基、叔戊基、1-甲基丁基、正己基、2-甲基戊基、3-甲基戊基、2,2-二甲基丁基、2,3-二甲基丁基、正庚基、2-甲基己基、3-甲基己基、2,2-二甲基戊基、2,3-二甲基戊基、2,4-二甲基戊基、3,3-二甲基戊基、3-乙基戊基、2,2,3-三甲基丁基、正辛基、异辛基、壬基、癸基、十一烷基、十二烷基、十三烷基、十四烷基、十五烷基、十六烷基、十七烷基、十八烷基、十九烷基、二十烷基等。
[0098]
通式(3-1)表示的化合物优选为下述通式(3-1-1)表示的化合物。x
1
是氢原子以外的选自于上述式(a)~(c)表示的组中的任意基团时,通过键合于该位置,x
1
作为给电子基团有效地发挥作用,吡啶环的n原子的亲核性有变得更好的倾向。
[0099]
[化8]
[0100][0101]
(式(3-1-1)中,x
1
表示与上述式(3-1)中的x
1
相同的意思。)
[0102]
当x
1
为上述式(c)表示的基团,r
33
以及r
34
相互键合形成环,r
33
或者r
34
中未直接键合的、上述烷基中的1个亚甲基被氧原子替换的情况下,通式(3-1)表示的化合物包括下述式(3-1-2)表示的4-吗啉基吡啶。
[0103]
[化9]
[0104][0105]
作为上述吡啶和吡啶衍生物,可以举例示出吡啶、上述4-吗啉基吡啶、n,n-二甲基-4-氨基吡啶、4-吡咯烷基吡啶和4-甲氧基吡啶作为优选物质。其中,通过使用4-吗啉基吡啶以及n,n-二甲基-4-氨基吡啶,单位时间内的酰胺的合成收率高,并且能够显著减少副反应物的生成,从这点来看特别优选上述两种物质。
[0106]
使用上述举例示出的吡啶以及吡啶衍生物时的阳离子活性物质是酰基吡啶鎓阳离子(酰基吡啶鎓物质)。酰基吡啶鎓物质具有亲电性高的特征。因此,即使是与后述亲核性低的胺的反应,也能够以非常快的速度进行反应,并且能够显著降低副反应物的生成。
[0107]
咪唑衍生物可以是咪唑的一个以上的氢原子被其它基团取代的物质,只要具有碱的性质即可,不受到特别限定,咪唑和咪唑衍生物优选为下述通式(3-2)所示的化合物。
[0108]
[化10]
[0109][0110]
(式中,r
35
和r
36
各自独立地为氢原子或烷基。)
[0111]
作为r
35
和r
36
的烷基,可以举出在r
31
、r
32
、r
33
以及r
34
的上述烷基中示例的烷基。
[0112]
作为上述咪唑和咪唑衍生物,可以举例示出咪唑以及n-甲基咪唑作为优选物质。
[0113]
另外,除了吡啶、吡啶衍生物、咪唑、咪唑衍生物以外,可以举例示出1,4-二氮杂双环[2,2,2]辛烷(dabco)作为优选物质。
[0114]
<工序3>
[0115]
工序3是使上述工序2中得到的上述阳离子活性物质与胺反应而制备酰胺的工序。
[0116]
实施方式的酰胺制备方法的工序3是使下述通式(4)表示的阳离子活性物质与下述通式(6)表示的胺反应,得到下述通式(7)表示的酰胺的工序。
[0117]
[化11]
[0118][0119]
(式中,r
1
、r
2
、r
3
以及r
4
是各自独立的氢原子或一价有机基团。)
[0120]
所述胺优选为氨基酸或氨基酸衍生物。
[0121]
作为氨基酸和氨基酸衍生物,可以举出在上述羧酸中例示的物质。
[0122]
当上述式(6)为氨基酸时,-r
3
和-r
4
例如可以是-h和-ch(r
0
)cooh。
[0123]
作为氨基酸衍生物的示例,可以举出官能团被保护基保护的保护氨基酸。作为被保护基保护的部位,可以举出选自于由氨基、羧基以及侧链组成的组中的任意一种以上的
部位。侧链中包含的官能团的1处或2处以上可以被保护基保护。在该工序3中,优选羧基和/或侧链的官能团被保护,以防止氨基以外的反应性官能团的反应。
[0124]
作为保护基的种类,不受到特别限定,可以根据被保护的官能团的种类适当选择。羧基的保护可以仅为中和成盐的形式,但通常以形成酯的形式进行保护。作为酯,除了甲基、乙基等烷基酯以外,举出苄基酯(有时简写为bn或bzl)等,但并不限于此。
[0125]
对于实施方式的酰胺制备方法,在工序3中使上述阳离子性活性物质与胺反应。在此,由于上述阳离子性活性物质的亲电性高,因此实施方式的酰胺制备方法具有反应速度不受胺的亲核性影响的优点。
[0126]
因此,实施方式的酰胺制备方法适合于与亲核性低的胺的反应。具体而言,亲核性低的胺可以是比18种氨基酸(从构成蛋白质并作为遗传信息编码的20种氨基酸中除去缬氨酸和异亮氨酸)的亲核性更低的胺,更具体而言,可以举例示出缬氨酸、异亮氨酸或n-烷基化的氨基酸或者上述物质的衍生物。n-烷基化的氨基酸可以是键合于α碳的氨基的1个或2个氢原子被烷基取代的氨基酸,优选1个氢原子被甲基取代的n-甲基氨基酸。这些亲核性低的胺以往难以在酸酐法中用于合成。然而,根据实施方式的酰胺制备方法,即使是以往难以在酸酐法中用于合成的亲核性低的胺也可以使用,在这一点上,实施方式的酰胺制备方法也是划时代的。
[0127]
在此,例如可以在实施例1所示的条件下进行酸酐法,使实施例1中生成的酸酐与想要求出亲核性的胺反应,根据其反应效率程度来求出胺的亲核性。
[0128]
在本实施方式中,只要考虑工序1~3的反应时各化合物的种类,根据目标反应适当地调节各化合物的使用量即可。
[0129]
活化的羧酸与胺的反应体系内的摩尔当量比(活化的羧酸∶胺)可以为10:1至1/10:1,也可以为5:1至1/5:1,还可以为3:1至1/3:1。活化的羧酸例如上述通式(4)表示的化合物。根据实施方式的酰胺制备方法,相对于活化的羧酸,即使与接近等当量的较少量胺反应的情况下,也能够高效率地制备酰胺。
[0130]
在本实施方式中,各工序的反应时间只要根据反应温度等其它条件适当地调节即可。作为一个例子,工序1的反应时间可以为0.05秒~30分钟,也可以为0.1秒~5分钟,还可以为0.5秒~30秒。在同时进行工序2和工序3的情况下,工序2和工序3的反应时间可以为1秒~60分钟,也可以为5秒~30分钟,还可以为1分钟~10分钟。
[0131]
在本实施方式中,工序1~3的反应时的温度(反应温度)只要根据工序1~3中使用的化合物的种类适当地调节即可。作为示例,反应温度优选为0~100℃的范围,更优选为20~50℃的范围。
[0132]
在本实施方式中,工序1~工序3的反应可以在溶剂的共存下进行。所述溶剂不受特别的限定,优选为不妨碍化合物的反应的溶剂,优选反应中所用原料的溶解性高的溶剂。例如,可以举出n,n-二甲基甲酰胺(dmf)、四氢呋喃(thf)、1,4-二氧六环等。
[0133]
在本实施方式中,在能够实现酰胺的生成的范围中,工序1~工序3的反应在反应体系内还可以包含不属于上述举例示出的化合物的其它化合物。
[0134]
在本实施方式中,工序1~工序3的反应可以分别进行,也可以同时进行。从更有效地抑制副反应物的生成的观点来看,优选同时进行工序2和工序3。
[0135]
在以上说明的实施方式的酰胺制备方法中,可以通过利用nmr、ir、质谱等解析得
到的光谱的测定、元素分析等,来确认产物的存在和结构。另外,根据需要可以对产物进行精制,可以通过蒸馏、提取、重结晶、柱层析等作为精制方法进行生成。
[0136]
根据实施方式的酰胺制备方法,能够非常高效率地制备酰胺。即使是在工序1中得到的酸酐,也处于作为活性物质接受亲核物质(胺)的状态。在本方法中,进一步在工序2中形成阳离子活性物质,相应于此开始与胺反应。由于此处生成的阳离子活性物质具有比酸酐显著更高的活性,所以能够以非常快的度进行反应,与以往方法相比,能够显著地抑制副产物的生成。进而,即使与在以往方法中反应困难的反应性低的胺也能够容易地进行反应。
[0137]
<肽的制备方法>
[0138]
在上述羧酸为氨基酸或氨基酸衍生物,并且上述胺为氨基酸或氨基酸衍生物的情况下,实施方式的酰胺制备方法能够合成肽或蛋白质。肽或蛋白质的制备方法包含在酰胺的制备方法中。
[0139]
将在上述工序3中得到的酰胺用作工序1中的羧酸,在工序1~3之后进一步重复进行工序1~3,由此能够使多肽链延伸。
[0140]
即,作为所述羧酸也包括多肽,作为实施方式的氨基酸或氨基酸衍生物(羧酸),也包括作为多肽构成单元的位于c末端的氨基酸或氨基酸衍生物(羧酸)。这样,实施方式的酰胺制备方法适合作为肽或蛋白质的制备方法。
[0141]
<流通系反应装置>
[0142]
实施方式的酰胺制备方法可以使用流通系反应装置进行实施。流通系反应装置可以举例示出具备以下部件的装置:输送流体的流路,所述流体包含实施方式的酰胺制备方法的反应中使用的原料或中间体;以及用于对所述流体进行混合的混合器。对于流通系反应装置的使用,例如,在流通系反应装置中至少可以进行上述工序3中与胺的反应,也可以在流通系反应装置中进行上述工序2以及工序3中与碱反应、与胺反应的反应,还可以在流通系反应装置中进行所述工序1~3中在使羧酸之间脱水缩合后与碱反应、与胺反应的反应。另外,实施方式的酰胺制备方法不限于使用流通系反应装置进行实施。例如也可以使用具有小容积和高速搅拌速度的批式容器(
バッチ
容器)。批式容器的混合部的体积可以为1~100ml,也可以为5~50ml。
[0143]
以下,参照图1对实施方式的流通系反应装置的方式以及使用该装置的实施方式的酰胺制备方法进行说明。
[0144]
图1是表示流通系反应装置1的概略构成的模式图。流通系反应装置1具备容纳第1液体的罐11、容纳第2液体的罐12、容纳第3液体的罐13。
[0145]
作为实例,第1液体包含第一羧酸和第二羧酸,第2液体包含光气或者在反应体系内分解后生成光气的光气等价物,第3液体包含碱和胺。作为更具体的实例,如图1所示,第1液体包含羧酸和diea,第2液体包含三光气,第3液体包含4-吗啉基吡啶和胺。
[0146]
对于流通系反应装置的使用,例如可以在流通系反应装置中至少进行第1液体和第2液体的混合物同第3液体的混合,进而也可以在流通系反应装置中进行第1液体同第2液体的混合。
[0147]
流通系反应装置1具备用于输送流体的流路f1、流路f2、流路f3、流路f4、流路f5。作为实例,流路的内径可以为0.1mm~10mm,也可以为0.3mm~8mm。流通系反应装置1具备用于混合流体的混合器31、混合器32。作为实例,混合器内部的流路的内径可以是0.1mm~
10mm,也可以是0.3mm~8mm。作为混合器,举出不具有驱动部的静态混合器(
スタティックミキサー
)。驱动部是指被赋予动力而运作的部分。
[0148]
上述“流路的内径”可以是在与流路长度方向垂直相交的方向上的流路截面中流路内部分(流体通过的部分)的直径。在流路内部分的形状不是正圆形的情况下,上述流路的内径可以是将上述流路内部分的形状以面积基准换算为正圆时的直径。
[0149]
作为实例,罐11、罐12、罐13、罐14、混合器31、混合器32以及流路f1、流路f2、流路f3、流路f4、流路f5由塑料或弹性体等树脂、玻璃材料、金属、陶瓷等形成。
[0150]
罐11与泵21连接,通过泵21的运转,罐11中容纳的第1液体在流路f1内移动并流入混合器31。罐12与泵22连接,通过泵22的运转,罐12中容纳的第2液体在流路f2内移动并流入混合器31。然后,第1液体和第2液体通过混合器31混合而成为第1混合液,送向流路f4。在该混合后的过程中,在第1液体中包含的羧酸之间发生脱水缩合,得到酸酐(酰胺制备方法的工序1)。得到的包含酸酐的第1混合液流入混合器32。
[0151]
另一方面,罐13与泵23连接,通过泵23的运转,罐13中容纳的液体在流路f3内移动并流入混合器32,与第1混合液混合而成为第2混合液,送向流路f5。在该混合后的过程中,在工序1中得到的酸酐与第3液体中包含的4-吗啉基吡啶反应而成为阳离子活性物质(酰胺制备方法的工序2),接下来,得到的阳离子活性物质与第3液体中包含的胺反应而得到酰胺(酰胺制备方法的工序3)。所制备的包含酰胺的第2混合液储存在罐14中。
[0152]
通过实施方式所述的流通系反应装置1,能够增大每单位体积的反应溶液进行热交换的面积。此外,反应时间可以通过流量、流路的长度来控制。因此,能够严格控制反应溶液,从而能够使不期望的副反应的进行得以最小化,能够提高目标产物的收率。
[0153]
在上述工序2中得到的阳离子活性物质由于活性度高,所以具有即使是反应性低的胺也能够使其反应的优点,另一方面,反应的控制变得重要。另外,即使是在工序1中得到的酸酐,活性度也足够高,因此反应的控制变得重要。
[0154]
根据实施方式的流通系反应装置1,使液体通过流路连续地流动,由此能够提高化合物碰撞的机会,能够以更高的效率进行反应,副反应的抑制也变得容易。例如,由于能够使在工序1中生成的酸酐立即与4-吗啉基吡啶(碱)反应,所以能够缩短酸酐处于活化状态的时间,能够降低发生异构化等副反应的概率。
[0155]
另外,在本实施方式的流通系反应装置中,举例示出了通过混合器对液体进行混合的方式,但是由于液体的混合可以仅通过流路彼此连通来实现,所以实施方式的流通系反应装置也可以不必具备混合器。
[0156]
如本文所示,实施方式的酰胺制备方法可以通过液相法实施。例如,目前主流的肽(酰胺)的制备方法是固相法,在固相上合成肽。另一方面,液相法适合于大规模的合成,由于分子的自由度提高,因此反应性也良好。液相法在与反应性低的胺的反应中也发挥效果。
[0157]
另外,在本实施方式所述的流通系反应装置中,将反应的5种化合物分开容纳在3个罐中,但例如也可以分别容纳在共计5个各自的罐中,依次混合。
[0158]
然而,如作为上述实施方式的第3液体所示,4-吗啉基吡啶(碱)和胺优选预先存在于相同液体中。即,工序2和工序3可以同时进行,由此,容易使工序2中产生的反应性高的阳离子性活性物质立即与目标胺反应,能够缩短阳离子性活性物质处于活化状态的时间,能够有效地抑制不期望的副反应物的生成。
[0159]
至此,参照化学式以及附图对本发明的实施方式进行了详细说明,但实施方式中的各结构以及它们的组合等只是实例,在不脱离本发明的主旨的范围内,可以进行结构的附加、省略、取代以及其它变更。此外,本发明不受各实施方式的限定,仅受权利要求的范围的限定。
[0160]
实施例
[0161]
以下示出实施例对本发明进行更详细的说明,但是本发明并不限于以下的实施例。
[0162]
<实施例1>本发明的酰胺制备方法
[0163]
[原料]
[0164]
对于用作羧酸的氨基酸,采用氨基被fmoc基保护、组氨酸侧链的用4-甲氧基苄氧基甲基(mbom)基保护的组氨酸,亦即fmoc-his(mbom)-oh(市售品)。对于用作胺的氨基酸,采用羧基被甲基保护、氨基被甲基化的苯丙氨酸,亦即h-mephe-ome(市售品)。
[0165]
[酸酰胺的流动合成]
[0166]
用作羧酸的氨基酸与用作胺的氨基酸进行偶联反应。偶联反应使用由ptfe制管(内径0.8mm,外径1.59mm)以及t字型混合器构成的流通系反应装置。反应前的溶液分成三种进行调制。将用作羧酸的fmoc-his(mbom)-oh和diea溶解于dmf中,得到第1溶液。将三光气溶解于thf中得到第2溶液。将用作胺的h-mephe-ome和4-吗啉基吡啶溶解于thf中,得到第3溶液。相对于h-mephe-ome为1.0,流通系反应装置中各物质的摩尔浓度比是:4-吗啉基吡啶为0.010,三光气为0.40,diea为3.0,fmoc-his(mbom)-oh为2.5。
[0167]
为了在流通系反应装置中进行偶联,首先,利用t字型混合器将第1溶液与第2溶液混合,通过在流通系反应装置中反应1秒钟,得到对称酸酐。其后立即利用新的t字型混合器将包含对称酸酐的反应溶液与第3溶液混合,在流通系反应装置中反应30秒,分配到试管中后反应约5分钟。这些反应全部在30℃下实施,将各反应前的溶液到达混合器前用于进行热交换的时间设定为20秒。各种溶液由注射泵流出。各泵的流量分别为:第1溶液2.0ml/min,第2溶液1.2ml/min,第3溶液2.0ml/min。
[0168]
以下示出实施例1中酰胺制备方法的工序1的反应。
[0169]
[化12]
[0170][0171]
[式中,r
h
表示组氨酸侧链(在本实施例中用保护基mbom保护)。]
[0172]
以下示出实施例1中酰胺制备方法的工序2的反应。
[0173]
[化13]
[0174][0175]
[式中,r
h
表示组氨酸侧链(在本实施例中用保护基mbom保护)。]
[0176]
以下示出实施例1中酰胺制备方法的工序3的反应。
[0177]
[化14]
[0178][0179]
[式中,r
h
表示组氨酸侧链(在本实施例中用保护基mbom保护),r
p
表示苯丙氨酸侧链。]
[0180]
[分析法]
[0181]
使用gpc分离目标产物,通过400mhz的h
1-nmr进行鉴定。
[0182]
异构化率的分析使用gc-ms进行。
[0183]
样品的调制如下进行。将得到的二肽的保护基除去后,将肽/氨基酸衍生物在重氢盐酸(重水素塩酸)中水解,用甲醇中的重氢化物将试样酯化,使试剂蒸发后,使用三氟乙酸酐或五氟丙酸酐将残留物酰基化。
[0184]
目标产物的收率由分离纯化的目标产物的重量算出。即,将胺的摩尔当量比作为1.0,由分离的二肽的重量计算胺偶联的比例。
[0185]
[结果]
[0186]
得到的二肽的nmr数据如下所示。
[0187]
1
h nmr(400mhz,cdcl
3
,主要异构体):δ7.78-6.85(m,20h),5.33-5.22(m,3h),4.79-4.74(m,1h),4.44-4.30(m,4h),4.15(t,j=6.8hz,1h),3.79(s,3h),3.72(s,3h),3.33(dd,j=5.5,14.5hz,1h),3.03(dd,j=6.8,14.5hz,1h),2.95-2.85(m,2h),2.67(s,3h).
[0188]
对反应后的产物进行分析的结果是,作为目标产物的二肽的偶联收率为84%,其中,his部位异构化的比例为1.1%。另外,未检测到异构化以外的副反应物。
[0189]
根据实施例1的方法,尽管羧酸与胺的摩尔浓度比为1∶2.5,也能够在5分钟的短时
间内实现80%以上的高的偶联收率。另外,目标产物中包含的差向异构体的生成率为1%左右,并且没有检测到其它副产物。
[0190]
<比较例1>混合酸酐法
[0191]
[原料]
[0192]
对于用作羧酸的氨基酸,采用氨基被fmoc基保护、侧链的π位被4-甲氧基苄氧基甲基(mbom)基保护的组氨酸,亦即fmoc-his(mbom)-oh。对于用作胺的氨基酸,采用羧基用甲基保护、氨基被甲基化的苯丙氨酸亦即h-mephe-ome。
[0193]
[酸酰胺的流动合成]
[0194]
用作羧酸的氨基酸与用作胺的氨基酸进行偶联反应。偶联反应使用了由ptfe制管(内径0.8mm,外径1.59mm)以及t字型混合器构成的流通系反应装置。反应前的溶液分成三种进行调制。将用作羧酸的fmoc-his(mbom)-oh、n-甲基吗啉和diea溶解于dmf中得到第1溶液。将氯甲酸异丙酯溶解于thf中得到第2溶液。将用作胺的h-mephe-ome和4-吗啉基吡啶溶解于thf中得到第3溶液。相对于h-mephe-ome为1.0,流通系反应装置中各物质的摩尔浓度比是:4-吗啉基吡啶为0.010,其它的fmoc-his(mbom)-oh、n-甲基吗啉、diea以及氯甲酸异丙酯均为1.0。
[0195]
为了在流通系反应装置中进行偶联,首先,利用t字型混合器将第1溶液与第2溶液混合,通过在流通系反应装置中反应20秒钟,得到混合酸酐。其后,立即使用新的t字型混合器将包含混合酸酐的反应溶液与第3溶液混合,在流通系反应装置中反应30秒,分配到试管中后反应约5分钟。这些反应全部在30℃下实施,作为在各反应前的溶液到达混合器前用于进行热交换的时间设定为20秒。各种溶液由注射泵流出。各泵的流量为:第1溶液1.2ml/min,第2溶液2.0ml/min,第3溶液2.0ml/min。
[0196]
[分析法]
[0197]
通过与实施例1相同的方法进行。
[0198]
另外,对于酯以及目标产物的分离,由于目标产物与酯具有同等程度的极性,所以使用gpc将酯与目标产物分离,利用400mhz的h
1-nmr进行鉴定。根据混合物的分离获取量以及用nmr得到的酯与目标产品物的峰面积的比率,计算出酯以及目标产物的收率。
[0199]
[结果]
[0200]
对反应后的产物进行分析的结果是,作为目标产物的二肽的偶联收率为26.4%。原料羧酸中的33.5%在作为副反应的酯化中被消耗。
[0201]
为了得到本来的目标产物,作为活化羧酸的酰基吡啶鎓物质(阳离子活性物质)应当受到来自胺的亲核进攻。但是,在比较例1的方法中,认为酰基吡啶鎓物质(阳离子活性物质)以与胺同等或更高的比例受到其平衡阴离子的亲核进攻。认为在与平衡阴离子的反应中得到的副反应物(即,酯)在同一反应中消耗33.5%的羧酸,其结果是,作为目标产物的二肽的收率降低到26.4%。
[0202]
附图标记说明
[0203]
1
…
流通系反应装置
[0204]
11、12、13、14
…
罐
[0205]
21、22、23
…
泵
[0206]
31、32
…
混合器
[0207]
f1、f2、f3、f4、f5
…
流路
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1