TREX1的调节剂的制作方法
trex1的调节剂
1.相关申请
2.本技术要求2018年12月6日提交的美国临时申请第62/776,301号的优先权,所述申请的全部内容以引用的方式并入本文中。
背景技术:
3.与非自身的先天免疫系统识别有关的癌症需要潜在免疫疗法,并且用以检测和防止潜在的危险。癌细胞在抗原上不同于其正常对应物,并发出类似于病毒感染的危险信号来警示免疫系统。这些信号,包括损伤相关分子模式(damp)和病原体相关分子模式(pamp),进一步激活了先天免疫系统,从而保护宿主免受多种威胁(《细胞与感染微生物学前沿(front.cell infect.microbiol.)》2012,2,168)。
4.异位表达的单股dna(ssdna)和双股dna(dsdna)为已知的pamp和/或damp,其由环状gmp
‑
amp合酶(cgas)(一种核酸感测器)来识别(《自然(nature)》2011,478,515
‑
518)。在感测到胞质dna后,cgas催化生成环状二核苷酸2
′
,3
′‑
cgamp,其为干扰素基因的er跨膜衔接蛋白刺激因子(sting)的有效第二信使和激活剂(《细胞报告(cell rep.)》2013,3,1355
‑
1361)。sting激活通过tbk1触发irf3的磷酸化,继而引起i型干扰素产生和干扰素刺激基因(isg)的激活;一种激活先天性免疫和启动适应性免疫的先决条件。因此,i型干扰素的产生构成了先天性免疫与适应性免疫之间的关键桥梁(《科学(science)》2013,341,903
‑
906)。
5.过量i型ifn可对宿主有害且诱导自身免疫性,因此,存在抑制i型ifn介导的免疫活化的负反馈机制。3
′
修复核酸外切酶i(trex1)是负责去除异位表达的ssdna和dsdna的3
′‑5′
dna核酸外切酶,且因此是cgas/sting途径的关键阻遏物(《美国国家科学院院刊(pnas)》2015,112,5117
‑
5122)。
6.i型干扰素和下游促炎性细胞因子反应对于免疫反应的发展和其有效性至关重要。i型干扰素增强树突状细胞和巨噬细胞吸收、处理、呈现和交叉呈现抗原至t细胞的能力,以及其通过引起共刺激分子(如cd40、cd80和cd86)的上调来刺激t细胞的能力(《实验医学杂志(j.exp.med.)》2011,208,2005
‑
2016)。i型干扰素还结合其自身的受体且激活有助于激活参与适应性免疫的细胞的干扰素反应性基因(《欧洲分子生物学学会报告(embo rep.)》2015,16,202
‑
212)。
7.从治疗的角度来看,i型干扰素和可诱导i型干扰素产生的化合物具有用于治疗人类癌症的潜力(《自然综述免疫学(nat.revimmunol.)》2015,15,405
‑
414)。干扰素可直接抑制人类肿瘤细胞增殖。另外,i型干扰素可通过触发来自先天性和适应性免疫系统两者的细胞的活化来增强抗肿瘤免疫性。重要的是,pd
‑
1阻断的抗肿瘤活性需要预先存在的瘤内t细胞。通过将冷肿瘤转变为热肿瘤并由此引发自发的抗肿瘤免疫,i型干扰素诱导疗法有可能扩大对抗pd
‑
1疗法有反应的患者群以及增强抗pd1疗法的有效性。
8.目前正在开发的诱导有效i型干扰素反应的疗法需要局部或瘤内施药以达到可接受的治疗指数。因此,仍需要具有全身递送和较低毒性的新药剂,以将i型ifn诱导疗法的益处扩展至没有外周治疗可及的病灶的患者。人类和小鼠遗传研究表明,trex1抑制可能适合
于全身递送途径,且因此trex1抑制性化合物可在抗肿瘤治疗领域中发挥重要作用。trex1是癌细胞对放射治疗作出反应的免疫原性有限的关键决定因素[《细胞生物学趋势(trends in cell biol.)》,2017,27(8),543
‑
4;《自然通讯(nature commun.)》,2017,8,15618]。trex1由遗传毒性应激诱导并参与神经胶质瘤和黑素瘤细胞对抗癌药物的保护[《生物化学与生物物理学报(biochim.biophys.acta)》,2013,1833,1832
‑
43]。stact
‑
trex1疗法在多个鼠类癌症模型中展示稳固的抗肿瘤功效[glickman等人,海报(poster)p235,第33届癌症免疫疗法学会年会(33
rd annual meeting of society for immunotherapy of cancer),washington dc,11月7
‑
11日,2018]。
技术实现要素:
[0009]
本文所描述的实施例大体上涉及用于治疗对trex1抑制剂有反应的疾病的药剂、化合物、组合物和方法。已在本发明中成功地合成对trex2抑制剂具有选择性的trex1抑制剂。
[0010]
本文提供具有式i的化合物:
[0011][0012]
和其药学上可接受的盐和组合物,其中a、r1、r2和n如本文所述。所公开的化合物和组合物调节trex1,并且适用于多种治疗应用,如治疗癌症。
附图说明
[0013]
图1a说明使用crispr进行的b16f10肿瘤细胞中的trex1的敲低实验的结果。
[0014]
图1b说明trex1减弱了b16f10肿瘤细胞中的cgas/sting途径的激活。
[0015]
图2:说明与亲本b16f10肿瘤相比,trex被沉默的肿瘤的体积更小。
[0016]
图3:展示trex1敲除b16f10肿瘤展现总体免疫细胞的显著增加。这反映肿瘤浸润cd4和cd8 t细胞以及浆细胞样树突状细胞(pdc)数目的增加。
[0017]
图4:展示某些本发明化合物于人类hct116结肠直肠癌细胞系中的活性。
具体实施方式
[0018]
1.化合物的一般描述
[0019]
在第一实施例中,本文提供式i化合物:
[0020][0021]
或其药学上可接受的盐,其中
[0022]
r1为氢、(c1‑
c4)烷基、卤基(c1‑
c4)烷基、3至4元环烷基、
‑
or
f
、
‑
sr
f
或
‑
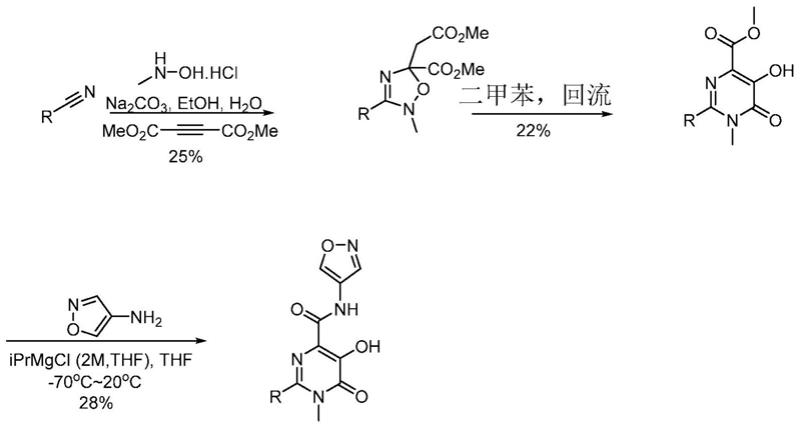
nr
e
r
f
;
[0023]
r2为氢、(c1‑
c4)烷基、卤基(c1‑
c4)烷基或3至4元环烷基;
[0024]
x为键、nr3、o、s或(c1‑
c4)亚烷基,其中所述(c1‑
c4)亚烷基任选地经1至2个选自r4的基团取代;
[0025]
r3为氢、(c1‑
c4)烷基、
‑
c(o)r
d
或
‑
c(s)r
d
;
[0026]
r4为卤基、(c1‑
c4)烷基、苯基、
‑
nhc(o)or
a
、
‑
nhc(s)or
a
、
‑
c(o)r
b
、
‑
nhc(o)nhr
g
、
‑
nhc(s)nhr
g
、
‑
nhs(o)2nhr
g
、
‑
c(s)r
b
、s(o)2r
c
、s(o)r
c
、
‑
c(o)or
d
、
‑
c(s)or
d
、
‑
c(o)nhr
e
、
‑
c(s)nhr
e
、
‑
nhc(o)r
d
、
‑
nhc(s)r
d
、
‑
or
e
、
‑
sr
e
、
‑
o(c1‑
c4)烷基or
e
或
‑
nr
e
r
f
,其中r4的所述苯基任选地经1或2个选自以下的基团取代:卤基、(c1‑
c4)烷基、卤基(c1‑
c4)烷基、(c1‑
c4)烷氧基和卤基(c1‑
c4)烷氧基;
[0027]
环a为苯基、5至6元杂芳基、4至7元杂环基或3至7元环烷基,其中的每一个任选地且独立地经1或2个选自r5的基团取代;
[0028]
r5为(c1‑
c4)烷基、卤基(c1‑
c4)烷基、卤基(c1‑
c4)烷氧基、卤基、苯基、
‑
nhc(o)or
a
、
‑
nhc(s)or
a
、
‑
c(o)r
b
,
‑
nhc(o)nhr
g
、
‑
nhc(s)nhr
g
、
‑
nhs(o)2nhr
g
、
‑
c(s)r
b
、s(o)2r
c
、s(o)r
c
、
‑
c(o)or
d
、
‑
c(s)or
d
、
‑
c(o)nr
e
r
f
、
‑
c(s)nhr
e
、
‑
nhc(o)r
d
、
‑
nhc(s)r
d
、
‑
or
e
、
‑
sr
e
、
‑
o(c1‑
c4)烷基or
e
、
‑
nr
e
r
f
、4至6元杂芳基或4至7元杂环基,其中r5的所述苯基任选地经1或2个选自r
g
的基团取代,r5的所述(c1‑
c4)烷基任选地经1或2个选自or
h
、
‑
nr
j
r
k
、苯基和5至6元杂芳基的基团取代,所述4至7元杂环基和4至6元杂芳基各自任选地且独立地经1或2个选自r
m
的基团取代,且其中r5中针对(c1‑
c4)烷基所列的所述任选的取代基的所述苯基和5至6元杂芳基各自任选地且独立地经1或2个选自r
g
的基团取代;
[0029]
r
g
、r
h
、r
j
、r
k
和r
m
各自独立地为氢、卤基、(c1‑
c4)烷基、卤基(c1‑
c4)烷基、(c1‑
c4)烷氧基、卤基(c1‑
c4)烷氧基、苯基、
‑
(c1‑
c4)烷基苯基、3至4元环烷基、4至6元杂芳基或4至7元杂环基,且其中r
g
、r
h
、r
j
和r
k
的所述4至7元杂环基进一步任选地经=o取代。
[0030]
r
a
、r
b
、r
c
、r
d
、r
e
和r
f
各自独立地为氢、卤基、(c1‑
c4)烷基、卤基(c1‑
c4)烷基、(c1‑
c4)烷氧基、卤基(c1‑
c4)烷氧基、苯基、3至4元环烷基、4至6元杂芳基或4至7元杂环基,其中i)r
a
、r
b
、r
c
、r
d
、r
e
和r
f
的所述(c1‑
c4)烷基任选地经1或2个选自苯基、
‑
or
h
、
‑
nr
j
r
k
的基团取代;ii)r
a
、r
b
、r
c
、r
d
、r
e
和r
f
的所述苯基、4至6元杂芳基和4至7元杂环基各自任选地且独立地经1或2个选自r
g
的基团取代,和iii)r
a
、r
b
、r
c
、r
d
、r
e
和r
f
的所述4至7元杂环基进一步任选地经=o取代。
[0031]
2.定义
[0032]
当用于描述可能有多个附接点的化学基团时,连字符(
‑
)表示这一基团与定义其
10393201。此基因编码人类细胞中的主要3
′‑
>5
′
dna核酸外切酶。所述蛋白质是一种非加工性核酸外切酶,可为人类dna聚合酶提供校对功能。它也是set复合物的一个组成部分,且用以在颗粒酶a介导的细胞死亡过程中迅速降解带切口dna的3
′
末端。缺乏功能性trex1的细胞展示内源性单股dna底物的慢性dna损伤检查点活化和核外积累。似乎trex1蛋白通常作用于由处理异常复制中间物产生的单链dna多核苷酸物质。trex1的此作用减弱dna损伤检查点信号传导且防止病理性免疫活化。trex1根据细胞内在抗病毒监测代谢内源性逆转录元件的逆转录单股dna,从而产生有效的i型ifn反应。trex1通过降解细胞质中的病毒cdna来帮助hiv
‑
1逃避胞质感测。
[0043]
术语“trex2”是指3
′
修复核酸外切酶2,其为在人体中由trex2基因编码的酶。此基因编码具有3
′
至5
′
核酸外切酶活性的核蛋白质。编码的蛋白质参与双股dna断裂修复,且可与dna聚合酶δ相互作用。具有此活性的酶参与dna复制、修复和重组。trex2为3
′‑
核酸外切酶,其主要在角化细胞中表达且有助于表皮对uvb诱导的dna损伤的反应。trex2生物化学和结构特性类似于trex1,尽管其不相同。两种蛋白质共享二聚结构且可在几乎相同的k
ca
值下在体外处理ssdna和dsdna底物。然而,与酶动力学、结构域和亚细胞分布有关的若干特征将trex2与trex1区分开来。与trex1相比,trex2对于体外dna底物呈现低10倍的亲和力。与trex1相比,trex2缺乏可介导蛋白质
‑
蛋白质相互作用的cooh
‑
末端域。trex2位于细胞质和细胞核两者中,而trex1发现于内质网中,且在颗粒酶a介导的细胞死亡期间或在dna损伤之后移动至细胞核。
[0044]
术语“受试者”和“患者”可互换使用,且意指需要治疗的哺乳动物,例如伴侣动物(例如狗、猫等)、农畜(例如母牛、猪、马、绵羊、山羊等)和实验动物(例如大鼠、小鼠、天竺鼠等)。通常,受试者是需要治疗的人。
[0045]
术语“抑制(inhibit/inhibition/inhibiting)”包括生物活性或过程的基线活性的降低。
[0046]
如本文所用,术语“治疗(treatment)”、“治疗(treat)”和“治疗(treating)”是指逆转、减轻如本文所述的疾病或病症或其一或多种症状,延迟其发作,或抑制其进展。在一些方面,治疗可在产生一或多种症状之后施用,即治疗性治疗。在其它方面,治疗可在不存在症状的情况下施用。举例来说,治疗可在症状发作之前向易感个体施用(例如根据症状病史和/或根据暴露于特定生物体或其它易感性因素),即防治性治疗。治疗还可在症状已消退之后继续,例如以延迟其复发。
[0047]
术语“药学上可接受的载剂”是指不会破坏与其一起调配的化合物的药理活性的无毒的载剂、佐剂或赋形剂。可用于本文所述组合物的药学上可接受的载剂、佐剂或赋形剂包括但不限于离子交换剂、氧化铝、硬脂酸铝、卵磷脂、血清蛋白如人血清白蛋白、缓冲物质如磷酸盐、甘氨酸、山梨酸、山梨酸钾、饱和植物脂肪酸的偏甘油酯混合物、水、盐或电解质,如硫酸鱼精蛋白、磷酸氢二钠、磷酸氢钾、氯化钠、锌盐、胶体二氧化硅、三硅酸镁、聚乙烯吡咯烷酮、纤维素类物质、聚乙二醇、羧甲基纤维素钠、聚丙烯酸酯、蜡、聚乙二醇
‑
聚氧丙烯嵌段聚合物、聚乙二醇和羊毛脂。
[0048]
供用于药品中,本文所述的化合物的盐是指无毒“药学上可接受的盐”。药学上可接受的盐形式包括药学上可接受的酸性/阴离子或碱性/阳离子盐。本文所述化合物的适合的药学上可接受的酸加成盐包括例如无机酸(如盐酸、氢溴酸、磷酸、硝酸和硫酸)和有机酸
(如乙酸、苯磺酸、苯甲酸、甲磺酸和对甲苯磺酸)的盐。具有酸性基团(如羧酸)的本传授内容的化合物可与药学上可接受的碱形成药学上可接受的盐。合适的药学上可接受的碱性盐包括例如铵盐、碱金属盐(如钠盐和钾盐)和碱土金属盐(如镁和钙盐)。具有季铵基团的化合物也含有抗衡阴离子,如氯离子、溴离子、碘离子、乙酸根、过氯酸根等。此类盐的其它实例包括盐酸盐、氢溴酸盐、硫酸盐、甲烷磺酸盐、硝酸盐、苯甲酸盐以及与氨基酸(如谷氨酸)的盐。
[0049]
术语“有效量”或“治疗有效量”是指将引发受试者的所需或有益生物或医疗反应的本文所述的化合物的量,例如0.01
‑
100毫克/千克体重/天之间的剂量。
[0050]
3.化合物
[0051]
在第二实施例中,本文提供式ii化合物:
[0052][0053]
或其药学上可接受的盐,其中所述变量如上文第一实施例中所描述。
[0054]
在第三实施例中,r2为式i或式ii化合物中的(c1‑
c4)烷基。
[0055]
在第四实施例中,本文提供式iii化合物:
[0056][0057]
或其药学上可接受的盐,其中所述变量如上文第一、第二或第三实施例中所描述。
[0058]
在第五实施例中,式i、式ii或式iii化合物中的环a为苯基、吡啶基、吡唑基、环丙基、环丁基、氮杂环丁基或哌啶基,其中的每一个任选地且独立地经一个或两个r5取代,其中其余的变量如上文针对第一、第二、第三或第四实施例所描述。或者,作为第五实施例的一部分,式i、式ii或式iii化合物中的环a为三唑基、吡咯烷基、二氮杂环庚烷基或哌嗪基,其中的每一个任选地且独立地经一个或两个r5取代,其中其余的变量如上文针对第一、第二、第三或第四实施例所描述。
[0059]
在第六实施例中,式i、式ii或式iii化合物中的r5为(c1‑
c4)烷基、卤基(c1‑
c4)烷基、卤基(c1‑
c4)烷氧基、卤基、苯基、
‑
nhc(o)or
a
、
‑
c(o)r
b
、s(o)2r
c
、s(o)r
c
、
‑
c(o)or
d
、
‑
c(o)nr
e
r
f
、
‑
nhc(o)r
d
、
‑
or
e
、
‑
o(c1‑
c4)烷基or
e
、
‑
nr
e
r
f
、4至6元杂芳基或4至7元杂环基,其中r5的所述苯基任选地经1或2个选自r
g
的基团取代,r5的所述(c1‑
c4)烷基任选地经1或2个选自
‑
or
h
、
‑
nr
j
r
k
、苯基和5至6元杂芳基的基团取代,所述4至6元杂芳基和4至7元杂环基各自任选
地且独立地经1或2个选自r
m
的基团取代,且其中r5中针对(c1‑
c4)烷基所列的所述任选的取代基的所述苯基和5至6元杂芳基各自任选地且独立地经1或2个选自r
g
的基团取代;ra为任选地经苯基取代的(c1‑
c4)烷基;r
b
为(c1‑
c4)烷基、苯基、5至6元杂芳基或4至7元杂环基,其中所述(c1‑
c4)烷基任选地经1或2个选自苯基、
‑
or
h
和
‑
nr
j
r
k
的基团取代,其中所述苯基、5至6元杂芳基和4至7元杂环基各自任选地且独立地经1或2个选自r
g
的基团取代,且其中所述4至7元杂环基进一步任选地经=o取代;各r
c
独立地为苯基或(c1‑
c4)烷基;各rd为氢或(c1‑
c4)烷基;各r
e
独立地为氢或任选地经or
h
取代的(c1‑
c4)烷基;各r
f
独立地为氢、(c1‑
c4)烷基、苯基、3至4元环烷基、4至6元杂芳基或5至6元杂环基,其中所述苯基、3至4元环烷基、4至6元杂芳基和5至6元杂环基各自任选地且独立地经r
g
取代;各r
g
独立地为(c1‑
c4)烷基、卤基(c1‑
c4)烷基、(c1‑
c4)烷氧基、卤基(c1‑
c4)烷氧基或卤基;各r
h
为氢、(c1‑
c4)烷基或
‑
(c1‑
c4)烷基苯基;各r
j
独立地为氢或(c1‑
c4)烷基;各r
k
独立地为氢、(c1‑
c4)烷基或3至4元环烷基;且各r
m
为(c1‑
c4)烷基,其中其余的变量如上文针对第一、第二、第三、第四或第五实施例所述。
[0060]
在第七实施例中,式i、式ii或式iii化合物中的r5为(c1‑
c4)烷基、卤基(c1‑
c4)烷基、卤基、苯基、
‑
nhc(o)or
a
、
‑
c(o)r
b
、s(o)2r
c
、
‑
c(o)or
d
、
‑
c(o)nr
e
r
f
、
‑
nhc(o)r
d
、
‑
o(c1‑
c4)烷基or
e
、
‑
nr
e
r
f
、4至6元杂芳基或5至6元杂环基,其中r5的所述(c1‑
c4)烷基任选地经
‑
or
h
、苯基或5至6元杂芳基取代,所述4至6元杂芳基和5至6元杂环基各自任选地且独立地经1或2个选自r
m
的基团取代,且其中r5中针对(c1‑
c4)烷基所列的所述任选的取代基的所述苯基和5至6元杂芳基各自任选地且独立地经1或2个选自r
g
的基团取代,其中其余的变量如上文针对第一、第二、第三、第四、第五或第六实施例所述。
[0061]
在第八实施例中,式i、式ii或式iii化合物中的r5为(c1‑
c4)烷基、卤基(c1‑
c4)烷基、卤基、苯基、
‑
nhc(o)or
a
、
‑
c(o)r
b
、s(o)2r
c
、
‑
c(o)or
d
、
‑
c(o)nr
e
r
f
、
‑
nhc(o)r
d
或
‑
o(c1‑
c4)烷基or
e
、
‑
nr
e
r
f
、吗啉基、哌嗪基或吡唑基,其中所述吗啉基、哌嗪基和吡唑基各自任选地经1或2个选自r
m
的基团取代,r5的所述(c1‑
c4)烷基任选地经
‑
or
h
、苯基、吡唑基、嘧啶基或吡啶基取代,且其中r5中针对(c1‑
c4)烷基所列的所述任选的取代基的所述苯基、吡唑基、嘧啶基和吡啶基各自任选地且独立地经1或2个选自r
g
的基团取代,其中其余的变量如上文针对第一、第二、第三、第四、第五、第六或第七实施例所述。
[0062]
在第九实施例中,式i、式ii或式iii化合物中的各r
f
独立地为氢、(c1‑
c4)烷基、苯基吡唑基、吡啶基、四氢吡喃基、哌啶基,其中所述苯基吡唑基、吡啶基、四氢吡喃基和哌啶基各自任选地且独立地经(c1‑
c4)烷基取代,其中其余的变量如上文针对第一、第二、第三、第四、第五、第六、第七或第八实施例所述。
[0063]
在第十实施例中,式i、式ii或式iii化合物中的各r
g
独立地为(c1‑
c4)烷基、(c1‑
c4)烷氧基或卤基,其中其余的变量如上文针对第一、第二、第三、第四、第五、第六、第七、第八或第九实施例所述。
[0064]
在第十一实施例中,式i、式ii或式iii化合物中的r
b
为(c1‑
c4)烷基、苯基、5至6元杂芳基或4至7元杂环基,其中r
b
的所述苯基和5至6元杂芳基各自任选地且独立地经1或2个选自卤基和(c1‑
c4)烷氧基的基团取代,其中r
b
的所述4至7元杂环基任选地经=o取代,且其中r
b
的所述(c1‑
c4)烷基任选地经1或2个选自苯基、
‑
or
h
和
‑
nr
j
r
k
的基团取代,其中其余的变量如上文针对第一、第二、第三、第四、第五、第六、第七、第八、第九或第十实施例所述。
[0065]
在第十二实施例中,式i、式ii或式iii化合物中的各r
k
独立地为氢或(c1‑
c4)烷基,
其中其余的变量如上文针对第一、第二、第三、第四、第五、第六、第七、第八、第九、第十或第十一实施例所述。
[0066]
在第十三实施例中,式i、式ii或式iii化合物中的r
b
为(c1
‑
c4)烷基、苯基、吡啶基、吡唑基、嘧啶基或哌啶基,其中r
b
的所述苯基、吡啶基、吡唑基和嘧啶基各自任选地且独立地经1或2个选自卤基和(c1‑
c4)烷氧基的基团取代,其中r
b
的所述(c1‑
c4)烷基任选地经1或2个选自苯基、oh和nme2的基团取代,且其中r
b
的所述哌啶基任选地经=o取代,其中其余的变量如上文针对第一、第二、第三、第四、第五、第六、第七、第八、第九、第十、第十一或第十二实施例所述。
[0067]
在第十四实施例中,式i、式ii或式iii化合物中的r3为氢,其中其余的变量如上文针对第一、第二、第三、第四、第五、第六、第七、第八、第九、第十、第十一、第十二或第十三实施例所述。
[0068]
在第十五实施例中,式i、式ii或式iii化合物中的r4为苯基或
‑
nhc(o)or
a
,其中其余的变量如上文针对第一、第二、第三、第四、第五、第六、第七、第八、第九、第十、第十一、第十二、第十三或第十四实施例所述。
[0069]
在第十六实施例中,式i、式ii或式iii化合物中的x为键或ch2,其中其余的变量如上文针对第一、第二、第三、第四、第五、第六、第七、第八、第九、第十、第十一、第十二或第十三实施例所述。
[0070]
本文还提供医药组合物,其包含1)具有式i的化合物:
[0071][0072]
或其药学上可接受的盐,其中变量如上文针对第一、第二、第三、第四、第五、第六、第七、第八、第九、第十或第十一实施例所述;和2)药学上可接受的载剂。
[0073]
具有式i的化合物进一步公开于范例中且包括于本公开中。包括其药学上可接受的盐以及中性形式。
[0074]
4.用途、调配和施用
[0075]
本文所述的化合物和组合物一般适用于调节trex1的活性。在一些方面,本文所述的化合物和药物组合物抑制trex1的活性。
[0076]
在一些方面,本文所述的化合物和药物组合物适用于治疗与trex1功能相关的病症。因此,本文提供治疗与trex1功能相关的病症的方法,其包含向有需要的受试者施用治疗有效量的本文所述的化合物或其药学上可接受的盐,或包含所公开的化合物或其药学上可接受的盐的药物组合物。还提供本文所述的化合物或其药学上可接受的盐或包含所公开的化合物或其药学上可接受的盐的药物组合物的用途,其用于制造供治疗与trex1功能相关的病症用的药剂。还提供本文所述的化合物或其药学上可接受的盐或包含所公开的化合物或其药学上可接受的盐的药物组合物,用于治疗与trex1相关的病症。
[0077]
在一些方面,本文所述的化合物和药物组合物适用于治疗癌症。
[0078]
在一些方面,由本文所述的化合物和药物组合物治疗的癌症选自结肠癌、胃癌、甲状腺癌、肺癌、白血病、胰腺癌、黑素瘤、多发性黑素瘤、脑癌、cns癌、肾癌、前列腺癌、卵巢癌、白血病和乳腺癌。
[0079]
在一些方面,由本文所述的化合物和药物组合物治疗的癌症选自肺癌、乳腺癌、胰腺癌、结肠直肠癌和黑素瘤。
[0080]
在某些方面,本文所述的药物组合物被调配成用于向需要此类组合物的患者施用。本文所述的药物组合物可经口、肠胃外、通过吸入喷雾、局部、经直肠、经鼻、经颊、经阴道或经由植入式贮器施用。如本文中所用,术语“肠胃外”包括皮下、静脉内、肌肉内、关节内、滑膜内、胸骨内、鞘内、肝内、病灶内和颅内注射或输注技术。在一些实施例中,所述组合物通过口服方式、腹膜内方式或静脉内方式施用。本文所述的药物组合物的无菌可注射形式可为水性或油性悬浮液。这些悬浮液可以根据所属领域中已知的技术使用合适的分散剂或湿润剂和悬浮剂来调配。
[0081]
在一些方面,经口施用药物组合物。
[0082]
用于任何特定患者的具体剂量和治疗方案将取决于各种因素,包括所采用的具体化合物的活性、年龄、体重、总体健康状况、性别、饮食、施用时间、排泄率、药物组合和治疗医生的判断以及被治疗的特定疾病的严重程度。组合物中的本文所述的化合物的量还将取决于药物组合物中的特定化合物。
[0083]
范例
[0084]
化学合成
[0085]
下面的代表性实例旨在帮助说明本公开,且其不旨在也不应解释为限制本发明的范围。
[0086]
一般合成方案
[0087][0088]2‑
(3
‑
溴
‑5‑
甲基苯基)
‑5‑
羟基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺
[0089][0090]
步骤1:合成3
‑
(3
‑
溴
‑5‑
甲基苯基)
‑5‑
(2
‑
甲氧基
‑2‑
氧代乙基)
‑2‑
甲基
‑
2,5
‑
二氢
‑
1,2,4
‑
噁二唑
‑5‑
甲酸甲酯
[0091][0092]
在室温下向sm(5.16g,26.341mmol,2当量)、n
‑
甲基羟胺盐酸盐(1.10g,13.170mmol,1.00当量)于h2o(25.00ml)、etoh(25.00ml)中的搅拌溶液中逐份添加na2co3(0.837g,7.902mmol,0.6当量)。在80℃下搅拌所得溶液2小时。直至反应混合物冷却至室温为止,逐滴添加丁
‑2‑
炔二酸1,4
‑
二甲酯(2.06g,14.488mmol,1.1当量)。接着,在室温下搅拌反应混合物0.5小时。所得溶液用3
×
30ml乙酸乙酯萃取,且有机层经合并,用盐水洗涤且在真空下浓缩。将残余物施加至具有乙酸乙酯/石油醚(1∶5)的硅胶柱上。这产生1.3g呈浅黄色油状的a1(25%产率)。esi
‑
ms m/z=385.0[m+h]+。mw计算值:384.0。1h nmr(400mhz,氯仿
‑
d)δ7.67(t,j=1.8hz,1h),7.49(s,2h),3.86(s,3h),3.73(s,3h),3.45(d,j=16.6hz,1h),3.19(s,3h),3.07(d,j=16.6hz,1h),2.38(d,j=0.8hz,3h)。
[0093]
使用与以上步骤中所述的那些条件类似的条件以及适当起始物质合成以下中间物。
[0094]
表1
[0095]
[0096]
[0097][0098]
步骤2:合成2
‑
(3
‑
溴
‑5‑
甲基苯基)
‑5‑
羟基
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酸甲酯b1
[0099][0100]
向100ml圆底烧瓶中置于3
‑
(3
‑
溴
‑5‑
甲基苯基)
‑5‑
(2
‑
甲氧基
‑2‑
氧代乙基)
‑2‑
甲基
‑
2,5
‑
二氢
‑
1,2,4
‑
噁二唑
‑5‑
甲酸甲酯(1.00g,2.596mmol,1.00当量)、二甲苯(20.00ml)。在145℃下搅拌所得溶液5小时。将反应混合物冷却至室温。通过过滤收集固体,用己烷洗涤。在减压下干燥固体。这产生210mg呈白色固体状的2
‑
(3
‑
溴
‑5‑
甲基苯基)
‑5‑
羟基
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酸甲酯(22%产率)。esi
‑
ms m/z=353.0[m+h]+。mw计算值:352.0
[0101]
使用与以上步骤中所述的那些条件类似的条件以及适当起始物质合成以下中间物。
[0102]
表2
[0103]
[0104]
[0105][0106]
步骤3:合成2
‑
(3
‑
溴
‑5‑
甲基苯基)
‑5‑
羟基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺(实例1)
[0107][0108]
向8ml密封管中放入2
‑
(3
‑
溴
‑5‑
甲基苯基)
‑5‑
羟基
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酸甲酯(100.00mg,0.283mmol,1.00当量)、thf(2.50ml)、1,2
‑
噁唑
‑4‑
胺(71.42mg,0.849mmol,3.00当量)。此后在
‑
70℃下搅拌下逐滴添加iprmgcl(2m,0.64ml,4.50当量)的溶液。在室温下搅拌所得溶液1小时。接着通过添加2ml饱和nh4cl(水溶液)来淬灭反应物。用hcl(4m)将溶液的ph值调节至3。所得溶液用3
×
5ml二氯甲烷萃取,且有机层经合并且在真空下浓缩。通过制备型hplc(ch3cn/h2o/fa)来纯化粗产物。这产生32.9mg呈白色固体状的2
‑
(3
‑
溴
‑5‑
甲基苯基)
‑5‑
羟基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺(28%产率)。esi
‑
ms m/z=405.0[m+h]+。mw计算值:404.0。1h nmr(300mhz,dmso
‑
d6)δ11.82(s,1h),10.95(s,1h),9.31(s,1h),8.87(s,1h),7.68(s,1h),7.62(s,1h),7.48(s,1h),3.30(s,3h),2.39(s,3h)。
[0109]
使用与以上步骤中所述的那些条件类似的条件以及适当起始物质合成以下实例。
[0110]
表3
[0111]
[0112][0113]
合成5
‑
羟基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑2‑
苯基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺(实例8)。
[0114][0115]
将5
‑
羟基
‑1‑
甲基
‑6‑
氧代基
‑2‑
苯基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酸甲酯(200mg,0.77mmol,1当量)和异噁唑
‑4‑
胺(0.096g,1.15mmol,1.5当量)溶解于甲苯(1ml)中且将混合物冷却至0℃。在0℃下向其中逐滴添加含2m三甲基铝的甲苯(0.768ml,1.53mmol,2.0当量)。接着将反应混合物在100℃下加热2小时,且接着用饱和碳酸氢钠溶液(0.2ml)淬灭,且向其中添加乙酸乙酯(10m1)。反应混合物接着经无水na2so4干燥且滤出。用乙酸乙酯(3
×
10m1)洗涤固体且在减压下浓缩合并的有机层。通过反相hplc来纯化粗产物,得到5
‑
羟基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑2‑
苯基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺,8(25mg,15%)。esi
‑
ms m/z=313.2[m+h]+。mw计算值:312.29。1hnmr(400mhz,dmso
‑
d6):δ9.13(s,1h),8.78(s,1h),7.42
‑
7.50(m,5h),3.22(s,3h)。
[0116]
方案:合成2
‑
氯
‑
n
‑
(异噁唑
‑4‑
基)
‑5‑
甲氧基
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺(c)
[0117][0118]
步骤1:合成2
‑
甲氧基
‑3‑
氧代丁二酸1,4
‑
二乙酯(1)
[0119][0120]
向用氮气惰性气氛吹扫和维持的1000ml 3颈圆底烧瓶中放入乙醇(600.0ml),在室温下添加乙醇钠(31.7g,0.470mmol,1.10当量)、乙二酸乙酯(68.00g,465.6mmol,1.10当量)。此后在室温下在搅拌下逐滴添加2
‑
甲氧基乙酸乙酯(50.00g,423.3mmol,1.00当量)。在35℃下搅拌所得溶液1整夜。在真空下浓缩所得混合物以去除大部分乙醇。在0℃下用氯化氢(1mol/l)将溶液的ph值调节至3。所得溶液用4
×
500 ml乙酸乙酯萃取,经无水硫酸钠干燥且在真空下浓缩。这产生90g(粗物质)呈棕色油状的2
‑
甲氧基
‑3‑
氧代丁二酸1,4
‑
二乙酯(1)。
[0121]
步骤2:合成2
‑
甲氧基
‑2‑
[(4e)
‑1‑
甲基
‑
2,5
‑
二氧代基咪唑烷
‑4‑
亚基]乙酸乙酯(2)
[0122][0123]
向2000ml圆底烧瓶中放入2
‑
甲氧基
‑3‑
氧代丁二酸1,4
‑
二乙酯(1)(90.00g,412.5mmol,1.00当量)、甲基脲(30.60g,412.5mmol,1.00当量)、乙酸(1200.0ml)、氯化氢(400.0ml,气体,4mol于二噁烷中)。在105℃下搅拌所得溶液3小时。在真空下浓缩所得混合物。用1
×
500 ml己烷洗涤所得混合物。这产生90g(粗物质)呈棕色固体状的2
‑
甲氧基
‑2‑
[(4e)
‑1‑
甲基
‑
2,5
‑
二氧代基咪唑烷
‑4‑
亚基]乙酸乙酯(2)。1h nmr(300mhz,氯仿
‑
d)δ8.75(s,1h),7.47(s,0.4h),5.09(s,2h),4.51
‑
4.30(m,3h),3.85(s,1h),3.84(s,3h),3.12(s,3h),3.07(s,1h),2.14(d,j=12.9hz,1h),1.45
‑
1.42(m,2h),1.42
‑
1.37(m,3h)。
[0124]
步骤3.合成2
‑
羟基
‑5‑
甲氧基
‑1‑
甲基
‑6‑
氧代嘧啶
‑4‑
甲酸(3)
[0125][0126]
向2000ml圆底烧瓶中放入2
‑
甲氧基
‑2‑
[(4e)
‑1‑
甲基
‑
2,5
‑
二氧代基咪唑烷
‑4‑
亚基]乙酸乙酯(2)(80.00g,350.6mmol,1.00当量)、氢氧化钾(1m于水中)(1400.0ml)。在105℃下搅拌所得溶液3小时。用水/冰浴将反应混合物冷却至0℃。在0℃下用氯化氢(12mol/l)将溶液的ph值调节至3,通过过滤收集固体且在真空中干燥沉淀物。这产生40g呈白色固体状的2
‑
羟基
‑5‑
甲氧基
‑1‑
甲基
‑6‑
氧代嘧啶
‑4‑
甲酸(3)(产率57%)。1h nmr(300mhz,dmso
‑
d6)δ14.35(s,1h),10.91(s,1h),3.68(s,3h),3.14(s,3h)。
[0127]
步骤4.合成2
‑
羟基
‑5‑
甲氧基
‑1‑
甲基
‑6‑
氧代嘧啶
‑4‑
甲酸乙酯(4)
[0128][0129]
向用氩气惰性气氛吹扫和维持的1000ml 3颈圆底烧瓶中放入2
‑
羟基
‑5‑
甲氧基
‑1‑
甲基
‑6‑
氧代嘧啶
‑4‑
甲酸(3)(20g,0.10mmol,1.00当量)、乙醇(400.0ml)。此后在0℃下在搅拌下逐滴添加乙酰氯(117.7g,1.500mmol,15.00当量)。将所得溶液加热至回流后维持1整夜。反应混合物用水/冰浴冷却。通过过滤收集固体。这产生16g呈白色固体状的2
‑
羟基
‑5‑
甲氧基
‑1‑
甲基
‑6‑
氧代嘧啶
‑4‑
甲酸乙酯(4)(产率:70%)。1hnmr(400mhz,dmso
‑
d6)δ11.08(s,1h),4.32(q,j=7.1hz,2h),3.70(s,3h),3.15(s,3h),1.31(t,j=7.1hz,3h)。
[0130]
步骤5.合成2
‑
氯
‑5‑
甲氧基
‑1‑
甲基
‑6‑
氧代嘧啶
‑4‑
甲酸乙酯(5)
[0131][0132]
向用氩气惰性气氛吹扫和维持的1l 3颈圆底烧瓶中放入2
‑
羟基
‑5‑
甲氧基
‑1‑
甲基
‑6‑
氧代嘧啶
‑4‑
甲酸乙酯(4)(16.0g,70.1mmol,1.00当量)、二甲基苯胺(1.20g,98.2mmol,1.40当量)、磷酰三氯(320.0ml)。在100℃下搅拌所得溶液1整夜。在真空下浓缩所得混合物。将残余物施加至具有乙酸乙酯/石油醚(1/10
‑
1/4)的硅胶柱上。这产生13.3g呈黄色固体状的2
‑
氯
‑5‑
甲氧基
‑1‑
甲基
‑6‑
氧代嘧啶
‑4‑
甲酸乙酯(5)(产率77.0%)。1hnmr(300mhz,dmso
‑
d6)δ4.32(q,j=7.1hz,2h),3.84(s,3h),3.54(s,3h),1.30(t,j=7.1hz,3h)。
[0133]
步骤6.合成2
‑
氯
‑5‑
甲氧基
‑1‑
甲基
‑
n
‑
(1,2
‑
噁唑
‑4‑
基)
‑6‑
氧代嘧啶
‑4‑
甲酰胺(6)
[0134][0135]
在室温下在氩气氛围下向2
‑
氯
‑5‑
甲氧基
‑1‑
甲基
‑6‑
氧代嘧啶
‑4‑
甲酸乙酯(5)(1.00g,4.05mmol,1.00当量)和1,2
‑
噁唑
‑4‑
胺(340.9mg,4.050mmol,1.00当量)于甲苯(15.0ml)中的搅拌溶液中添加三甲基铝(2mol于甲苯中)(4.1ml,8.10mmol,2.00当量)。将所得溶液在80℃下用微波辐射搅拌15分钟。将反应混合物在0℃下用水/冰淬灭。所得溶液用3
×
40ml乙酸乙酯萃取,合并的有机层用盐水(1
×
30ml)洗涤,经无水硫酸钠干燥且在真空下浓缩。将残余物施加至具有乙酸乙酯/石油醚(1/10
‑
1/1)的硅胶柱上。这产生650mg呈淡黄色固体状的2
‑
氯
‑5‑
甲氧基
‑1‑
甲基
‑
n
‑
(1,2
‑
噁唑
‑4‑
基)
‑6‑
氧代嘧啶
‑4‑
甲酰胺(6)(产率53.0%)。esi
‑
ms m/z=285.2[m+h]
+
.mw计算值:284.21h nmr(300mhz,dmso
‑
d6)δ10.84(s,1h),9.28(s,1h),8.78(s,1h),3.86(s,3h),3.62(s,3h)。
[0136]
使用与以上步骤中所述的那些条件类似的条件以及适当起始物质合成以下实例和中间物。
[0137]
表4
[0138]
[0139]
[0140][0141]
使用与上文所述的那些条件类似的条件以及适当起始物质合成以下中间物。
[0142]
表5
[0143][0144]
合成2
‑
(3
‑
溴
‑2‑
(2
‑
羟基乙氧基)苯基)
‑5‑
羟基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺(实例15)。
[0145][0146]
在室温下向40ml小瓶中添加2
‑
(3
‑
溴
‑2‑
(2
‑
((叔丁基二甲基硅烷基)氧基)乙氧基)苯基)
‑5‑
羟基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺(300mg,
0.531mmol,1.0当量)和thf(6.0ml)。在0℃下在2分钟内向以上混合物中逐滴添加tbaf(1m)/ch3cooh=1∶1(1.20ml,2.0当量)。在室温下再搅拌所得混合物5小时。用乙酸乙酯(1
×
12ml)萃取所得混合物。用乙酸乙酯(3
×
10ml)进一步萃取水相。合并的有机层用盐水(2
×
7ml)洗涤,经无水硫酸钠干燥。在过滤后,减压浓缩滤液。通过制备型hplc(乙腈/甲酸/水)纯化粗产物(300mg),得到70mg呈白色固体状的2
‑
(3
‑
溴
‑2‑
(2
‑
羟基乙氧基)苯基)
‑5‑
羟基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺15,产率为29%。esi
‑
ms m/z=451.0[m+h]+。mw计算值:450.0。1h nmr(300mhz,dmso
‑
d6)δ11.87(s,1h),10.99(s,1h),9.29(s,1h),8.86(s,1h),7.85(d,j=8.1hz,1h),7.56(d,j=7.6hz,1h),7.28(t,j=7.8hz,1h),4.63(t,j=5.4hz,1h),4.01
‑
3.97(m,1h),3.76
‑
3.70(m,1h),3.52
‑
3.39(m,2h),3.23(s,3h)。
[0147]
使用与以上步骤中所述的那些条件类似的条件以及适当起始物质合成以下实例和中间物。
[0148]
表6
[0149]
[0150]
[0151]
[0152]
[0153]
[0154]
[0155]
[0156]
[0157]
[0158]
[0159]
[0160]
[0161]
[0162][0163]
[0164]
合成3
‑
(5
‑
乙氧基
‑4‑
(异噁唑
‑4‑
基氨甲酰基)
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑2‑
基)哌啶
‑1‑
甲酸叔丁酯(异构体a1):
[0165][0166]
步骤1:2
‑
(1
‑
(叔丁氧基羰基)
‑
1,2,5,6
‑
四氢吡啶
‑3‑
基)
‑5‑
乙氧基
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酸乙酯:
[0167]
将2
‑
氯
‑5‑
乙氧基
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酸乙酯(2g,7.67mmol)、5
‑
(4,4,5,5
‑
四甲基
‑
1,3,2
‑
二氧杂硼杂环戊
‑2‑
基)
‑
3,6
‑
二氢吡啶
‑
1(2h)
‑
甲酸叔丁酯(2.96g,9.59mmol)和碳酸钠(1.62g,15.34mmol)于二噁烷/水的溶液(4∶1,50ml)中的混合物用氩气脱气20分钟。添加pdcl2(dppf)(0.561g,0.76mmol)且再继续脱气10分钟。在90℃下加热反应混合物4小时。反应完成后,将反应混合物溶解于水(100ml)中且用乙酸乙酯(2
×
50ml)萃取。合并的有机层用盐水(30ml)洗涤,经无水na2so4干燥且在减压下浓缩。粗产物通过柱色谱纯化,获得呈固体状的标题化合物(2.57g,82%)。lcms:计算值408.3;esi
‑
ms m/z=408.61[m+h]
+1
h nmr(400mhz,dmso
‑
d6):δ1.23
‑
1.31(m,6h),1.43(s,9h),2.25
‑
2.40(m,2h),3.44(s,3h),3.45
‑
3.50(m,2h),4.05
‑
4.10(m,2h),4.16(q,j=7.2hz,2h),4.31(q,j=7.2hz,2h),6.26(bs,1h)。
[0168]
步骤2:2
‑
(1
‑
(叔丁氧基羰基)哌啶
‑3‑
基)
‑5‑
乙氧基
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酸乙酯:
[0169]
向在甲醇∶乙酸乙酯的溶液(1∶3,54ml)中溶解并搅拌的2
‑
(1
‑
(叔丁氧基羰基)
‑
1,2,5,6
‑
四氢吡啶
‑3‑
基)
‑5‑
乙氧基
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酸乙酯(2.57g,6.30mmol)的混合物中添加10%pd/c(0.67g,具有50%水分)。在室温下在氢气氛围下搅拌反应物3.5小时。反应完成后,将反应混合物通过硅藻土床过滤且用乙酸乙酯(2
×
50ml)洗涤。在减压下浓缩滤液。获得的粗产物通过柱色谱纯化,得到标题化合物(2.35g,91%)。lcms:计算值409.3,esi
‑
ms m/z=410.4[m+h]
+
[0170]
步骤3:2
‑
(1
‑
(叔丁氧基羰基)哌啶
‑3‑
基)
‑5‑
乙氧基
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酸:
[0171]
在室温下向2
‑
(1
‑
(叔丁氧基羰基)哌啶
‑3‑
基)
‑5‑
乙氧基
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酸乙酯(1.9g,4.64mmol)于甲醇∶thf∶h2o的混合物(1∶1∶1,30ml)中的搅拌溶液中添加naoh(0.222g,5.56mmol)。在室温下搅拌所得反应混合物3小时。反应完成后,将反应物在真空下浓缩。将反应混合物溶解于水(10ml)中且用1n hcl(ph
‑
6)酸化。用含10%甲醇的二氯甲烷(2
×
30ml)萃取产物。合并的有机层用盐水(30ml)洗涤且在真空下蒸发,得到粗标题化合物(1.67g,94%),其不经进一步纯化即用于下一步骤中。lcms:计算值381.3;esi
‑
ms m/z=382.5[m+h]
+
[0172]
步骤4:3
‑
(5
‑
乙氧基
‑4‑
(异噁唑
‑4‑
基氨甲酰基)
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑2‑
基)哌啶
‑1‑
甲酸叔丁酯:
[0173]
在0℃下向2
‑
(1
‑
(叔丁氧基羰基)哌啶
‑3‑
基)
‑5‑
乙氧基
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酸(1.67g,4.37mmol)于无水dmf(16ml)中的搅拌溶液中添加hatu(2.49g,6.56mmol)。在室温下搅拌反应混合物1小时。接着在室温下添加含异噁唑
‑4‑
胺(0.441g,5.25mmol)的dmf(0.5ml)和dipea(1.5ml,8.75mmol),且将反应混合物搅拌3小时。反应完成后,将反应混合物用水(20ml)稀释且用乙酸乙酯(2
×
30ml)萃取。合并的有机层经无水na2so4干燥且在减压下浓缩。通过combiflash柱色谱来纯化粗产物,得到呈外消旋混合物形式的标题化合物(1.67g,85%)。通过使用手性制备型hplc来进行外消旋纯化合物(1.2g)的手性拆分,得到500mg的每种对映异构体。lcms:计算值447.3;esi
‑
ms m/z=448.60[m+h]
+
手性hplc:chiralpak ad
‑
h;含30%甲醇的液体co2+0.1%dea。fr
‑
1(异构体
‑
a1):r
t
=7.66min;fr
‑
2(异构体
‑
a2):r
t
=8.81min;a1:1h nmr(400mhz,dmso
‑
d6):δ1.21
‑
1.25(m,3h),1.41(s,9h),1.46
‑
1.57(m,2h),1.71
‑
1.74(m,2h),2.02
‑
2.04(m,1h),2.76
‑
2.98(m,2h),2.99
‑
3.15(m,1h),3.57(s,3h),3.85
‑
4.15(m,1h),4.10
‑
4.15(m,2h),8.78(s,1h),9.29(s,1h),10.56(s,1h)。a2:1h nmr(400mhz,dmso
‑
d6):δ1.21
‑
1.29(m,3h),1.41(s,9h),1.46
‑
1.57(m,2h),1.71
‑
1.74(m,2h),2.02
‑
2.04(m,1h),2.78
‑
2.98(m,2h),2.99
‑
3.15(m,1h),3.57(s,3h),3.85
‑
4.15(m,1h),4.10
‑
4.15(m,2h),8.78(s,1h),9.29(s,1h),10.57(s,1h)。
[0174]
合成1
‑
((2
‑
((r)
‑1‑
(3,4
‑
二氯苯甲酰基)哌啶
‑3‑
基)
‑4‑
(异噁唑
‑4‑
基氨甲酰基)
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑5‑
基)氧基)乙基碳酸乙酯。(实例131)
[0175][0176]
步骤1:(r)
‑5‑
乙氧基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑2‑
(哌啶
‑3‑
基)
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺盐酸盐:
[0177]
在0℃下向(r)
‑3‑
(5
‑
乙氧基
‑4‑
(异噁唑
‑4‑
基氨甲酰基)
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑2‑
基)哌啶
‑1‑
甲酸叔丁酯(0.20g,0.44mmol)于二氯甲烷(1.32ml)和甲醇(0.66ml)的混合物中的搅拌溶液中逐滴添加含4m hcl的二噁烷(1ml)。在室温下搅拌反应混合物1小时且升温至40℃后维持10分钟。将反应混合物浓缩至干燥且与甲醇共沸两次,得到呈固体状的标题化合物(0.17g,99%),其不经进一步纯化即用于下一步骤中。lcms:计算值347.3,esi
‑
ms m/z=348.48[m+h]
+
[0178]
步骤2:(r)
‑2‑
(1
‑
(3,4
‑
二氯苯甲酰基)哌啶
‑3‑
基)
‑5‑
乙氧基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺:
[0179]
在0℃下向3,4
‑
二氯苯甲酸(0.101g,0.53mmol)于无水dmf(1.7m1)中的搅拌溶液中添加hatu(0.252g,0.66mmol)。在室温下搅拌反应混合物1小时。接着在室温下添加(r)
‑5‑
乙氧基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑2‑
(哌啶
‑3‑
基)
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺盐酸盐(0.170g,0.44mmol)和dipea(0.25ml,1.33mmol),且搅拌反应混合物3小时。完成后,将反应物用水(15ml)稀释且用乙酸乙酯(2
×
20ml)萃取。合并的有机层用冷饱和碳酸氢钠(20ml)洗涤,经无水na2so4干燥且在减压下浓缩。通过柱色谱纯化粗产物,得到呈固体状的标题化合物(0.21g,89%)。lcms:计算值519.3,esi
‑
ms m/z=520.27[m+h]
+
。1h nmr(400mhz,dmso
‑
d6):δ1.18
‑
1.32(m,3h),1.55
‑
1.70(m,2h),1.72
‑
1.85(m,2h),2.07
‑
2.10(m,1h),3.15
‑
3.25(m,2h),3.58(s,3h),3.66
‑
3.70(m,1h),4.07
‑
4.12(m,2h),4.47
‑
4.49(m,1h),7.40
‑
7.42(m,1h),7.70
‑
7.73(m,2h),8.74(s,1h,),9.27(s,1h),10.52(s,1h)。
[0180]
步骤3:(r)
‑2‑
(1
‑
(3,4
‑
二氯苯甲酰基)哌啶
‑3‑
基)
‑5‑
羟基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺:
[0181]
将(r)
‑2‑
(1
‑
(3,4
‑
二氯苯甲酰基)哌啶
‑3‑
基)
‑5‑
乙氧基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺(0.608g,1.16mmol)的搅拌溶液溶解于二氯甲烷(36ml)中。将所得溶液冷却至0℃且逐滴添加含1m bbr3的二氯甲烷(4.67ml,4.67mmol)。将反应混合物升温至室温且搅拌5小时。完成后,将反应混合物浓缩,且与二氯甲烷(2
×
6ml)共沸。将残余物冷却至0℃且缓慢添加甲醇(5ml)。将反应混合物在真空中浓缩至干燥。残余物与甲醇(2
×
6ml)共沸,得到粗化合物。将粗化合物负载于硅藻土上,且通过使用乙腈和0.1%甲酸/水的反相柱色谱纯化,得到实例131(0.246g,42%)。lcms:计算值491.3,esi
‑
ms m/z=492.3[m+h]
+
。1h nmr(400mhz,dmso
‑
d6):δ1.44
‑
1.62(m,2h),1.80
‑
1.95(m,2h),2.07
‑
2.10(m,1h),3.15
‑
3.25(m,2h),3.57(s,3h),3.60
‑
3.70(m,1h),4.34
‑
4.52(m,1h),7.40
‑
7.42(m,1h),7.70
‑
7.74(m,2h),8.84
‑
8.90(m,1h,),9.30(s,1h),10.42
‑
10.55(m,1h),11.48(s,1h)。
[0182]
使用与以上步骤中所述的那些条件类似的条件以及适当起始物质合成以下实例和中间物。
[0183]
表7
[0184][0185]
[0186]
[0187][0188]
合成2
‑
(1
‑
(3,4
‑
二氯苯甲酰基)
‑3‑
甲基哌啶
‑3‑
基)
‑5‑
羟基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺.(实例130)
[0189][0190]
步骤1:3
‑
氰基
‑3‑
甲基哌啶
‑1‑
甲酸叔丁酯:
[0191]
在
‑
78℃下,向3
‑
氰基哌啶
‑1‑
甲酸叔丁酯(5.0g,24mmol)于无水thf(100ml)中的搅拌溶液中添加lihmds(30.9ml,1m于thf中,30.9mmol)。在
‑
78℃下搅拌反应混合物30分钟。接着在相同温度下添加mei(2.22ml,35.7mmol)。在室温下搅拌反应混合物16小时。反应完成后,将反应混合物导入冰冷水(100ml)的混合物中且用乙酸乙酯(2
×
150ml)萃取。合并的有机层用盐水(50ml)洗涤,经无水na2so4干燥且在减压下浓缩。通过硅胶色谱纯化所得残余物,得到纯标题化合物(3.5g,65%)1hnmr(400mhz,dmso
‑
d6):δ1.35(s,3h),1.43(s,9h),1.66
‑
1.70(m,1h),1.78
‑
1.81(m,2h),1.87
‑
2.10(m,1h),2.80(s,2h),3.90
‑
4.20(m,2h)。
[0192]
步骤2:3
‑
甲基哌啶
‑3‑
甲腈tfa盐。
[0193]
在0℃下向3
‑
氰基
‑3‑
甲基哌啶
‑1‑
甲酸叔丁酯(3.5g,15.6mmol)于二氯甲烷(70ml)中的搅拌溶液中逐滴添加tfa(12.7ml)。在室温下搅拌所得混合物3小时。在减压下浓缩反应混合物,得到粗化合物。用二乙醚湿磨粗化合物,得到呈tfa盐形式的标题化合物
(3.0g,81%)。1hnmr(400mhz,dmso
‑
d6):δ1.40(s,3h),1.63
‑
1.69(m,2h),1.76
‑
1.81(m,1h),1.91
‑
2.06(m,1h),2.82
‑
2.89(m,1h),3.01
‑
3.09(m,1h),3.19
‑
3.22(m,1h),3.56
‑
3.59(m,1h),9.20(bs,1h)。
[0194]
步骤3:1
‑
(3,4
‑
二氯苯甲酰基)
‑3‑
甲基哌啶
‑3‑
甲腈。
[0195]
在室温下向3,4
‑
二氯苯甲酸(2.88g,15.1mmol)于无水dmf(15ml)中的搅拌溶液中添加hatu(7.18g,18.9mmol)。在室温下搅拌反应混合物1小时。在室温下逐滴添加dipea(7ml,38mmol),接着添加3
‑
甲基哌啶
‑3‑
甲腈tfa盐(3.0g,12.6mmol)。在室温下搅拌反应混合物3小时。反应完成后,反应混合物用水(30ml)稀释,且水层用乙酸乙酯(2
×
200ml)萃取。合并的有机层用盐水(50ml)洗涤,经无水na2so4干燥且在减压下浓缩。通过柱色谱纯化所得残余物,得到纯标题化合物(2.8g,56%)。lcms:计算值296.3,esi
‑
ms m/z=297.2[m+h]+。
[0196]
步骤4:1
‑
(3,4
‑
二氯苯甲酰基)
‑
n
‑
羟基
‑3‑
甲基哌啶
‑3‑
甲脒:
[0197]
在0℃下向1
‑
(3,4
‑
二氯苯甲酰基)
‑3‑
甲基哌啶
‑3‑
甲腈(4g,13.5mmol)、羟胺盐酸盐(2.80g,40.4mmol)于乙醇(100ml)中的混合物中缓慢添加三乙胺(5.8ml,40.4mmol)。将所得反应混合物在70℃下加热16小时,接着浓缩,用水(100ml)稀释且用含10%甲醇的二氯甲烷(2
×
150ml)萃取。合并的有机层经无水na2so4干燥且在减压下浓缩,获得呈固体状的标题化合物(5.0g,69%)。粗产物不进一步纯化直接用于下一步骤。
[0198]
lcms:计算值329.3,esi
‑
ms m/z=330.4[m+h]+。
[0199]
步骤5:2
‑
((e)
‑1‑
(3,4
‑
二氯苯甲酰基)
‑
n
′‑
羟基
‑3‑
甲基哌啶
‑3‑
甲脒基)顺丁烯二酸二甲酯:
[0200]
在0℃下向1
‑
(3,4
‑
二氯苯甲酰基)
‑
n
‑
羟基
‑3‑
甲基哌啶
‑3‑
甲脒(5.0g,15mmol)于氯仿(100ml)中的搅拌溶液中逐滴添加乙炔二甲酸二甲酯(3.32g,23.4mmol),且在室温下搅拌反应混合物过夜。反应物通过tlc监测且接着浓缩,得到粗产物,其通过硅胶柱色谱纯化,获得呈固体状的纯标题化合物(2.4g,33%)。lcms:计算值471.2,esi
‑
ms m/z=472.3[m+h]+。
[0201]
步骤6:2
‑
(1
‑
(3,4
‑
二氯苯甲酰基)
‑3‑
甲基哌啶
‑3‑
基)
‑5‑
羟基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酸甲酯。
[0202]
将2
‑
((e)
‑1‑
(叔丁氧基羰基)
‑
n
′‑
羟基哌啶
‑3‑
甲脒基)顺丁烯二酸二甲酯(2.0g,4.2mmol)于二甲苯(15ml)中的溶液在160℃下在微波中加热10分钟。反应混合物经浓缩且用二乙醚和己烷湿磨以获得固体。将粗残余物负载于硅藻土上且通过使用乙腈和0.1%甲酸/水的反相柱色谱纯化,得到纯标题化合物(0.40g,21%)。lcms:lcms:计算值439.4,esi
‑
ms m/z=440.5[m+h]+。1h nmr(400mhz,dmso
‑
d6):δ1.10(s,2h),1.27(s,3h),1.58
‑
1.75(m,3h),2.00
‑
2.16(m,1h),3.79(s,3h),3.60
‑
3.64(m,1h),4.01(bs,1h),7.26(d,j=8.4,1h),7.54(s,1h),7.67(d,j=2.0hz,1h),10.36(s,1h),12.40
‑
12.65(m,1h)。
[0203]
步骤7:2
‑
(1
‑
(3,4
‑
二氯苯甲酰基)
‑3‑
甲基哌啶
‑3‑
基)
‑5‑
羟基
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酸甲酯。
[0204]
将2
‑
(1
‑
(3,4
‑
二氯苯甲酰基)
‑3‑
甲基哌啶
‑3‑
基)
‑5‑
羟基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酸甲酯(0.200g,0.454mmol)的混合物溶解于dmso(50ml)中且将其冷却至0℃。在0℃下向其中逐滴添加甲醇镁溶液(1.30ml,0.908mmol,6至10%于甲醇中)。在室温下搅拌反应混合物1小时。浓缩反应混合物以去除过量甲醇。将反应混合物冷却至0℃且逐滴添加mei
(0.116ml,1.81mmol)。将反应混合物在室温下搅拌16小时,接着冷却至10℃且用1n hcl缓慢淬灭。用乙酸乙酯(2
×
200ml)萃取产物。合并的有机层经无水na25o4干燥且在减压下浓缩。将粗残余物负载于硅藻土上且通过使用乙腈和0.1%甲酸/水的反相柱色谱纯化,得到纯标题化合物(0.20g,97%)。lcms:计算值453.4,esi
‑
ms m/z=454.2[m+h]+。1hnmr(400mhz,dmso
‑
d6):δ1.39(s,3h),1.59(bs,1h),1.75(bs,1h),2.01
‑
2.07(m,2h),3.15
‑
3.25(m,1h),3.32
‑
3.52(m,1h),3.63(s,3h),3.79(s,3h),3.83
‑
3.86(m,1h),3.99
‑
4.02(m,1h),7.30(d,j=8.4hz,1h),7.55(s,1h),7.69(d,j=2.0hz,1h),10.32(s,1h)。
[0205]
步骤8:2
‑
(1
‑
(3,4
‑
二氯苯甲酰基)
‑3‑
甲基哌啶
‑3‑
基)
‑5‑
甲氧基
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酸甲酯:
[0206]
在室温下向2
‑
(1
‑
(3,4
‑
二氯苯甲酰基)
‑3‑
甲基哌啶
‑3‑
基)
‑5‑
羟基
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酸甲酯(0.20g,0.44mmol)于dmf(1ml)中的搅拌溶液中添加k2co3(0.121g,0.875mmol),接着添加mei(0.042ml,0.660mmol)。在室温下搅拌反应混合物4小时。反应完成后,将其用水(20ml)稀释且用乙酸乙酯(2
×
20ml)萃取水层。合并的有机层用盐水(10ml)洗涤,经无水na2so4干燥且在减压下浓缩。通过柱色谱纯化所得残余物,得到纯标题化合物(0.14g,67%)。lcms:计算值467.4,esi
‑
ms m/z=468.3[m+h]
+
[0207]
步骤9:2
‑
(1
‑
(3,4
‑
二氯苯甲酰基)
‑3‑
甲基哌啶
‑3‑
基)
‑5‑
甲氧基
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酸:
[0208]
在室温下向2
‑
(1
‑
(3,4
‑
二氯苯甲酰基)
‑3‑
甲基哌啶
‑3‑
基)
‑5‑
甲氧基
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酸甲酯(0.140g,0.298mmol)于甲醇∶thf∶水的混合物(1∶1∶1,6ml)中的搅拌溶液中添加naoh(0.014g,0.36mmol)。在室温下搅拌反应混合物3小时。反应完成后,在减压下浓缩反应混合物,用冰冷水(10ml)稀释且用乙酸乙酯(2
×
10ml)萃取。合并的水层在0℃下用1n hc1酸化且用含10%甲醇的二氯甲烷(2
×
30ml)萃取。合并的有机层用盐水(20ml)洗涤,经无水na2so4干燥且在减压下浓缩,得到粗标题化合物(0.130g,96%)。lcms:计算值455.2,esi
‑
ms m/z=456.6[m+h]
+
[0209]
步骤10:2
‑
(1
‑
(3,4
‑
二氯苯甲酰基)
‑3‑
甲基哌啶
‑3‑
基)
‑
n
‑
(异噁唑
‑4‑
基)
‑5‑
甲氧基
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺。
[0210]
在室温下向2
‑
(1
‑
(3,4
‑
二氯苯甲酰基)
‑3‑
甲基哌啶
‑3‑
基)
‑5‑
甲氧基
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酸(0.140g,0.308mmol)于无水dmf(1.4ml)中的搅拌溶液中添加hatu(0.176g,0.462mmol)。在室温下搅拌反应混合物1小时,且接着在室温下添加dipea(0.17ml,0.99mmol)和异噁唑
‑4‑
胺(0.038g,0.46mmol)。在室温下搅拌反应混合物3小时,接着用水(20ml)稀释且用乙酸乙酯(2
×
20ml)萃取。合并的有机层用盐水(10ml)洗涤,经无水na2so4干燥且在减压下浓缩。通过柱色谱纯化所得残余物,得到呈固体状的纯标题化合物(0.135g,84%)。lcms:计算值519.2,esi
‑
ms m/z=520.4[m+h]+。1h nmr(400mhz,dmso
‑
d6):δ1.16
‑
1.19(m,3h),1.19
‑
1.23(m,2h),1.42
‑
1.56(m,2h),3.32
‑
3.52(m,2h)3.62(s,3h),4.01
‑
4.02(m,1h),4.03(s,3h),4.70
‑
4.77(m,1h),7.25
‑
7.27(m,1h),7.59
‑
7.67(m,2h),8.74(s,1h),9.27(s,1h),10.58(bs,1h)。手性hplc:chiralpak ad
‑
h;含30%(meoh)的液态co2+0.1%dea:fr
‑
1(异构体
‑
1):r
t
=9.39;fr
‑
2(异构体
‑
2):r
t
=14.82。
[0211]
步骤11:2
‑
(1
‑
(3,4
‑
二氯苯甲酰基)
‑3‑
甲基哌啶
‑3‑
基)
‑5‑
羟基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺。
[0212]
将溶解于二氯甲烷(2ml)中的2
‑
(1
‑
(3,4
‑
二氯苯甲酰基)
‑3‑
甲基哌啶
‑3‑
基)
‑
n
‑
(异噁唑
‑4‑
基)
‑5‑
甲氧基
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺fr
‑
1(0.030g,0.057mmol)的搅拌溶液冷却至0℃且逐滴添加含1m bbr3的二氯甲烷(0.28ml,0.29mmol)。接着在室温下搅拌反应混合物3小时。反应完成后,且接着在真空下浓缩。添加甲醇(1ml)且搅拌30分钟且蒸发。将残余物与甲醇(2
×
1ml)共沸,得到粗化合物(120mg)。将粗残余物负载于硅藻土上且通过使用乙腈和0.1%甲酸/水的反相柱色谱纯化,得到实例130(0.015g,51%)。lcms:计算值505.2,esi
‑
ms m/z=506.3[m+h]+。1h nmr(400mhz,dmso
‑
d6):δ1.35
‑
1.45(m,3h),1.46
‑
1.60(m,2h),1.70
‑
1.80(m,2h),3.15
‑
3.18(m,2h),3.38
‑
3.44(m,2h),3.61(s,3h),7.22
‑
7.24(m,1h),7.59
‑
7.64(m,2h),8.73(s,1h),9.23(s,1h),10.75(bs,1h),11.58(bs,1h)。
[0213]
合成5
‑
羟基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑2‑
(1
‑
苯乙基哌啶
‑3‑
基)
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺(实例129)
[0214][0215]
步骤1:5
‑
乙氧基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑2‑
(哌啶
‑3‑
基)
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺盐酸盐:
[0216]
在0℃下向3
‑
(5
‑
乙氧基
‑4‑
(异噁唑
‑4‑
基氨甲酰基)
‑1‑
甲基
‑6‑
氧代基
‑
1,6
‑
二氢嘧啶
‑2‑
基)哌啶
‑1‑
甲酸叔丁酯[来自a1的合成](0.35g,0.78mmol)于二氯甲烷(3ml)和甲醇(0.5ml)的混合物中的搅拌溶液中逐滴添加含4m hc1的二噁烷(3ml)。在室温下搅拌反应混合物1小时且升温至40℃后维持10分钟。将反应混合物浓缩至干燥且与甲醇共沸两次,得到呈固体状的标题化合物(0.290g,97%),其不经进一步纯化即用于下一步骤。lcms:计算值347.5;esi
‑
ms m/z=348.5[m+h]
+
[0217]
步骤2:5
‑
乙氧基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑2‑
(1
‑
苯乙基哌啶
‑3‑
基)
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺:
[0218]
向5
‑
乙氧基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑2‑
(哌啶
‑3‑
基)
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺盐酸盐(0.050g,0.130mmol)于dmf(0.5ml)中的搅拌溶液中添加三乙胺(0.055ml,0.391mmol)且在室温下搅拌30分钟。添加(2
‑
溴乙基)苯(0.024g,0.130mmol)且在室温下搅拌反应混合物4小时,接着浓缩。所获得的粗产物用己烷(5ml)湿磨且在真空下干燥,得到标题化合物(0.050g)。粗标题化合物不经进一步纯化即用于下一步骤中。lcms:
计算值451.3;esi
‑
ms m/z=452.4[m+h]
+
[0219]
步骤3:5
‑
羟基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑2‑
(1
‑
苯乙基哌啶
‑3‑
基)
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺:
[0220]
将5
‑
乙氧基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑2‑
(1
‑
苯乙基哌啶
‑3‑
基)
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺(0.05g,0.110mmol)的混合物溶解于二氯甲烷(1ml)中。将所得溶液冷却至0℃且逐滴添加含1m bbr3的二氯甲烷(0.22ml,0.22mmol)。在室温下搅拌反应混合物5小时,接着添加饱和碳酸氢钠(0.5ml)且进一步用含10%甲醇的二氯甲烷稀释。将残余物经na2so4干燥且浓缩至干燥。将粗残余物负载于硅藻土上且通过使用乙腈和0.1%甲酸/水的反相柱色谱纯化,得到纯标题化合物(0.01g,21%)。lcms:计算值423.4:esi
‑
ms m/z=424.5[m+h]
+
[0221]1h nmr(400mhz,dmso
‑
d6):δ1.60
‑
1.80(m,4h),1.85
‑
1.95(m,1h),2.66
‑
2.70(m,2h),2.70
‑
3.02(m,6h),3.48(s,3h),7.18
‑
7.28(m,5h),8.77(s,ih,),9.13(s,1h),10.25(bs,1h),11.50(bs,1h)。
[0222]
使用与以上步骤中所述的那些条件类似的条件以及适当起始物质合成以下实例和中间物。
[0223]
表8
[0224][0225]
合成5
‑
羟基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑2‑
(哌啶
‑1‑
基)
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺(实例28)
[0226][0227]
步骤1:
[0228]
将c6(200mg,0.769mmol)、哌啶(0.76ml,7.69mmol)和dmso的混合物在110℃下加热1小时。通过tlc监测反应进程且在消耗c6后将其冷却至室温。将反应混合物倒入冰水(25m1)的混合物中且充分搅拌15分钟。将固体材料滤出,用水(3
×
10ml)洗涤且在真空下充分干燥,得到呈固体状的纯产物(172mg,72%)。esi
‑
ms m/z=310.38[m+h]
+
mw计算值:309.371h nmr(400mhz,dmso
‑
d6):δ4.00(q,2h),4.26(q,2h),3.38(s,3h),3.04
‑
3.06(m,4h),1.57
‑
1.62(m,6h),1.20
‑
1.30(m,6h)。
[0229]
步骤2:在0℃下向5
‑
乙氧基
‑1‑
甲基
‑6‑
氧代基
‑2‑
(哌啶
‑1‑
基)
‑
1,6
‑
二氢嘧啶
‑4‑
甲酸乙酯(100mg,0.32mmol)、异噁唑
‑4‑
胺(35.3mg,0.42mmol)于甲苯(1ml)中的搅拌溶液中添加含2m三甲基铝的甲苯(0.6ml,0.64mmol)。将反应混合物在80℃下在微波照射下加热30分钟。通过tlc确认反应完成且将反应物倒入冰冷水的混合物中且用乙酸乙酯(2
×
10m1)萃取。有机层用盐水(2
×
10ml)洗涤。合并的有机层经na2so4干燥且在真空下蒸发,得到呈浅棕色固体状的粗化合物(104mg)。粗物质不经任何纯化即用于下一步骤中。esi
‑
ms m/z=348.5[m+h]
+
mw计算值:347.3
[0230]
步骤3:将5
‑
乙氧基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑2‑
(哌啶
‑1‑
基)
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺(100mg,0.28mmol)溶解于二氯甲烷(1ml)中。将所得溶液冷却至0℃且逐滴添加含1m bbr3的二氯甲烷(0.5ml,0.57mmol)。在室温下搅拌反应混合物2小时。通过tlc监测反应(含10%甲醇的二氯甲烷)。完成后,在真空下去除溶剂。将冰冷水添加至残余物中,且过滤固体以获得粗产物(83mg)。粗产物通过rp
‑
hplc进一步纯化,得到实例28,5
‑
羟基
‑
n
‑
(异噁唑
‑4‑
基)
‑1‑
甲基
‑6‑
氧代基
‑2‑
(哌啶
‑1‑
基)
‑
1,6
‑
二氢嘧啶
‑4‑
甲酰胺产物(10mg,0.032mmol,11%)。esi
‑
ms m/z=320.3[m+h]
+
mw计算值:319.31h nmr(400mhz,dmso
‑
d6):δ11.19(s,1h),10.54(s,1h),9.31(s,1h),8.92(s,1h),3.43(s,3h),3.08(s,4h),1.59
‑
1.67(m,6h)。
[0231]
使用与以上步骤中所述的那些条件类似的条件以及适当起始物质合成以下实例和中间物。
[0232]
表9
[0233]
[0234]
[0235]
[0236][0237]
生物化学分析
[0238]
1.沉默肿瘤细胞中的trex1
[0239]
在感测胞质dna和随后的i型ifn产生后,cgas/sting途径的活化可在肿瘤细胞和先天性免疫细胞,特别是树突状细胞中发生。为了评估trex1是否通过一种充分描述的冷同基因型肿瘤模型来抑制i型ifn的产生,所述模型在通过sting激动剂激活i型ifn后会经历免疫介导的排斥反应,使用crispr在b16f10肿瘤细胞中敲低了trex1(图1a)。相对于亲本肿
瘤细胞,通过肿瘤细胞的dna转染进行的胞质dna积累使得trex1敲除b16f10细胞的ifnβ产量增加了约5倍,表明trex1减弱了b16f10肿瘤细胞中cgas/sting途径的激活(图1b)。
[0240]
2.trex1胜任型和缺陷型b16f10肿瘤细胞在体内的生长
[0241]
评估了trex1胜任型和缺陷型b16f10肿瘤细胞在体内的生长。在c57bl/6j小鼠的右侧腹上皮下接种300,000个亲本或trex1敲除b16f10肿瘤细胞。每周收集体重两次,且每周进行肿瘤测量两到三次,在肿瘤变得可测量时开始且在研究的剩余持续时间内持续。与亲本b16f10肿瘤相比,trex1被沉默的肿瘤呈现显著更小的体积(图2)。
[0242]
在研究终止后的第19天收集肿瘤,并消化成单细胞悬浮液,以使得能够进行肿瘤浸润的免疫群体的流式细胞术定量。发现trex1敲除b16f10肿瘤展现总体免疫细胞的显著增加,其反映肿瘤浸润cd4和cd8 t细胞以及浆细胞样树突状细胞(pdc)数量的增加(图3)。已知pdc在诱导抗原特异性抗肿瘤免疫反应中起核心作用,而已知t细胞是小鼠和人类抗肿瘤功效的主要效应物。因此,缺乏trex1的肿瘤的免疫浸润的深刻变化表明,对后者肿瘤生长的抑制至少部分是免疫介导的。
[0243]
trex1生物化学分析
[0244]
通过荧光分析评估化合物效力,所述荧光分析测量在相对股上具有荧光团
‑
淬灭剂对的定制dsdna底物的降解。dsdna的降解会释放游离荧光团以产生荧光信号。确切地说,将反应缓冲液(50mm tris(ph 7.4),150mm nacl,2mm dtt,0.1mg/ml bsa,0.01%(v/v)tween
‑
20和100mm mgcl2中的7.5μl n末端his
‑
tev标记的全长人类trex1(在大肠杆菌中表达且内部纯化)添加至384孔black proxiplate plus(perkin elmer),其已含有改变浓度的化合物(150nl),作为dmso中的10点剂量反应。向其中添加7.5μl含dsdna底物(股a:5
′
tex615/gctagg cag 3
′
;股b:5
′
ctg cctagc/iabrqsp(integrated dna technologies))的反应缓冲液。最终浓度为含有1.0%dmso(v/v)的反应缓冲液中的150pm trex1、60nm dsdna底物。在室温下25分钟之后,通过添加5μl终止缓冲液(与反应缓冲液加200mm edta相同)来淬灭反应物。淬灭的反应物中的最终浓度为20μl体积内的112.5pm trex1、45nm dna和50mm edta。在室温下培育5分钟后,在激光源envision(perkin
‑
elmer)中读取板,在用570nm光激发后测量615nm处的荧光。通过使用非线性最小二乘四参数拟合和genedata或graphpad prism(graphpad software,inc.)比较615nm处测量的荧光相对于用终止缓冲液预淬灭的对照孔(100%抑制)和无抑制剂(0%抑制)对照的比率来计算ic
50
值。
[0245]
trex2生物化学分析
[0246]
通过荧光分析评估化合物效力,所述荧光分析测量在相对股上具有荧光团
‑
淬灭剂对的定制dsdna底物的降解。dsdna的降解会释放游离荧光团以产生荧光信号。确切地说,将反应缓冲液(50mm tris(ph 7.4),150mm nacl,2mm dtt,0.1mg/mlbsa,0.01%(v/v)tween
‑
20和100mm mgcl2)中的7.5μl n末端his
‑
tev标记的人类trex2(残基m44
‑
a279,在大肠杆菌中表达且内部纯化)添加至384孔black proxiplate plus(perkin elmer),其已含有改变浓度的化合物(150nl),作为dmso中的10点剂量反应。向其中添加7.5μl含dsdna底物(股a:5
′
tex615/gctagg cag 3
′
;股b:5
′
ctg cctagc/iabrqsp(idt))的反应缓冲液。最终浓度为含有1.0%dmso(v/v)的反应缓冲液中的2.5nm trex2、60nm dsdna底物。在室温下25分钟之后,通过添加5μl终止缓冲液(与反应缓冲液加200mm edta相同)来淬灭反应物。淬灭的反应混合物中的最终浓度为20μl体积内的1.875pm trex2、45nm dna和50mm edta。在室温
下培育5分钟后,在激光源envision(perkin
‑
elmer)中读取板,在用570nm光激发后测量615nm处的荧光。通过使用非线性最小二乘四参数拟合和genedata或graphpadprism(graphpad software,inc.)比较615nm处测量的荧光相对于用终止缓冲液预淬灭的对照孔(100%抑制)和无抑制剂(0%抑制)对照的比率来计算ic
50
值。
[0247]
结果展示于表1中。trex1 ic
a0
:a=<0.1μm;b=0.1至1μm;c=1至10μm;d=>10μm。trex2 ic
a0
:a=<1μm,b=1至10μm,c=10至100μm,d=>100μm。
[0248]
表10
[0249]
[0250]
[0251]
[0252][0253]
hct116细胞分析
[0254]
hct116双细胞(invivogen,san diego,ca,usa)来源于人类hct116结肠直肠癌细胞系。已选择用于稳定整合seap和荧光素酶报告基因的细胞,其表达分别受nf
‑
kb/ap1和stat1/stat2的5个串联反应元件控制。细胞系用于通过测量培养基中分泌的lucia荧光素酶的活性来监测i型干扰素诱导和后续信号传导。
[0255]
将hct116细胞在96孔板中以40,000个细胞/孔接种于补充有10%fbs和25mmhepes(ph 7.2
‑
7.5)的100μl dmem中。过夜沉降后,将细胞用trex1i处理4小时(最大dmso分数为0.1%),随后用脂染胺(lipofectamine)ltx(thermofisher,grand island,ny,usa)根据产品手册建议对1μg/mlpbr322/bstni限制消化物(new england biolabs,ipswich,ma,usa)进行转染。简单来说,将脂染胺ltx(0.35μl/孔)稀释于optimem(5μl/孔)中。将pbr322/bstni(100ng/孔)稀释于optimem(5μl/孔)中,随后添加plus试剂(0.1μl/100ng dna)。在室温下培育5分钟之后,将dna混合物与稀释的脂染胺ltx逐滴混合。再培育10分钟后,将转染混合物(10μl/孔)添加至细胞。将细胞在37c下维持48小时,随后自细胞培养基监测lucia荧光素酶活性。
[0256]
虽然我们已经描述了多个实施例,但显而易见的是,可以改变我们的基础实例以提供利用本发明的化合物和方法的其它实施例。因此,应了解,本发明范围应该由所附权利要求书而不是所举例表示的特定实施例来定义。
[0257]
在整个本技术中引用的所有参考文献(包括文献参考、授予的专利、公开的专利申请以及共同待决专利申请)的内容由此以全文引用的方式明确地并入本文中。除非另外定义,否则本文所使用的所有技术和科学术语与所属领域的一般技术人员通常已知的含义一致。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1