一种高效率的联吡啶钌类染料敏化剂及其制备方法和应用与流程
本发明涉及太阳能电池技术领域,具体涉及一种高效率的联吡啶钌类染料敏化剂及其制备方法和应用。
背景技术:
染料敏化太阳能电池(dye-sensitizedsolarcells,dsscs)是第三代新型太阳能光伏技术,自1991年瑞士洛桑高等工业学院的m.
典型的染料敏化太阳能电池包括导电玻璃、纳米半导体电极、染料敏化剂、电解质和对电极等,形成一种三明治式的结构。此类电池的核心思想是将光的吸收过程和电子收集过程分开,即分别由染料敏化剂和纳米半导体基底分别来完成,因此染料敏化剂在其中起着至关重要的作用。染料敏化剂起到了扩展器件吸收光谱范围的重要作用,这种敏化思想使得器件的吸收光谱拓展到了可见光区乃至近红外区,极大地提高了电池器件的光捕获能力。经过二十多年的发展,研究者们已经开发出了一大批优秀的染料。目前来说,用于dsscs器件的高效染料敏化剂主要分为三大类:联吡啶钌类敏化剂、金属卟啉类敏化剂和纯有机类敏化剂。
技术实现要素:
本发目的在于提供一种制备方法简单、产率较高、高效率的染料敏化剂。
本发明是通过如下技术方案实现的:
一种高效率的联吡啶钌类染料敏化剂,其分子结构式如式(a)
所示:
结构式(a)所示染料敏化剂以r1、r2基团和硫氰酸根作为电子供体端,以联吡啶二甲酸作为电子受体端兼锚固基团。
一种高效率的联吡啶钌类染料敏化剂的制备方法,包括如下步骤:
(1)将5-醛基-2-噻吩硼酸、碳链修饰的3-溴吩噻嗪、无机碱加入有机溶剂中,在惰性气体保护下加入催化剂并加热,反应完毕后冷却至室温,除去溶剂,然后萃取,对有机相干燥、过滤、旋蒸,经柱层析法纯化得到化合物(1);
(2)将化合物(1)、1,10-邻二氮杂菲-5,6-二酮、乙酸铵加入有机溶剂中,在惰性气体保护下将反应液加热,反应完毕后冷却至室温,向反应液中加水并过滤,然后重结晶得到化合物(2);
(3)冰浴条件下将化合物(2)加入有机溶剂中,加入强碱并搅拌,然后加入
(4)惰性气体保护下,将二氯双(4-甲基异丙基苯基)钌(ii)、化合物(3)加入有机溶剂中并加热反应,接着加入联吡啶双羧酸并加热反应,然后加入硫氰酸盐并加热反应,反应停止后,加水淬灭反应并过滤,然后先柱层析法纯化,再重结晶得到化合物(a)。
进一步,步骤(1)中所述5-醛基-2-噻吩硼酸、所述碳链修饰的3-溴吩噻嗪、所述无机碱和所述催化剂之间的摩尔当量比为1:(0.6-1):(3-5):(0.05-0.1),所述碳链修饰的3-溴吩噻嗪其碳链具有4-10个碳原子,所述加热温度为90-100℃,所述催化剂为pd(pph3)4或pd(dppf)cl2,所述无机碱为碳酸钠、碳酸钾、磷酸钾中任一种或几种,萃取剂为乙酸乙酯、二氯甲烷、三氯甲烷中任一种或几种。
进一步,步骤(2)中所述化合物(1)、所述1,10-邻二氮杂菲-5,6-二酮和所述乙酸铵之间的摩尔当量比为1:(0.9-1.1):(10-30),所述加热温度为100-140℃。
进一步,步骤(3)中所述化合物(2)、所述强碱和所述
进一步,步骤(4)中所述二氯双(4-甲基异丙基苯基)钌(ii)、所述化合物(3)、所述联吡啶双羧酸和所述硫氰酸盐之间摩尔当量比为1:(1.8-2):(1.8-2):(10-40),所述加入有机溶剂后的加热温度为80-120℃、所述加入联吡啶双羧酸后的加热温度为120-160℃、所述加入硫氰酸盐后的加热温度为120-160℃,所述反应时间均为4-8小时,所述硫氰酸盐为硫氰酸铵、硫氰酸钾、硫氰酸钠中任一种或几种。
进一步,步骤(1)中所述柱层析法选用具砂板层析柱作为固定相,填料为200-300目的硅胶,选用石油醚-乙酸乙酯为流动相,石油醚与乙酸乙酯的体积比为10:1;步骤(4)中所述柱层析法选用具砂板层析柱作为固定相,填料为200-300目的硅胶,选用甲醇-二氯甲烷为流动相,甲醇与二氯甲烷的体积比为1:20。
进一步,步骤(1)、步骤(2)、步骤(3)、步骤(4)中所述有机溶剂为n,n-二甲基甲酰胺、四氢呋喃、1,4-二氧六环、醋酸中任一种或几种。
一种高效率的联吡啶钌类染料敏化剂的应用,该染料敏化剂应用于太阳能电池中。
本发明的有益效果:
本发所得染料具有优异的光子捕获能力,吸收光谱响应达到了近700nm,单色-光电响应范围达到了800nm,并且在400-600nm范围内的单色光电转换效率均超过了70%。组装出的太阳能电池器件转换效率达到了9.17%,相同条件下明星染料n719的转换效率为8.08%。
附图说明
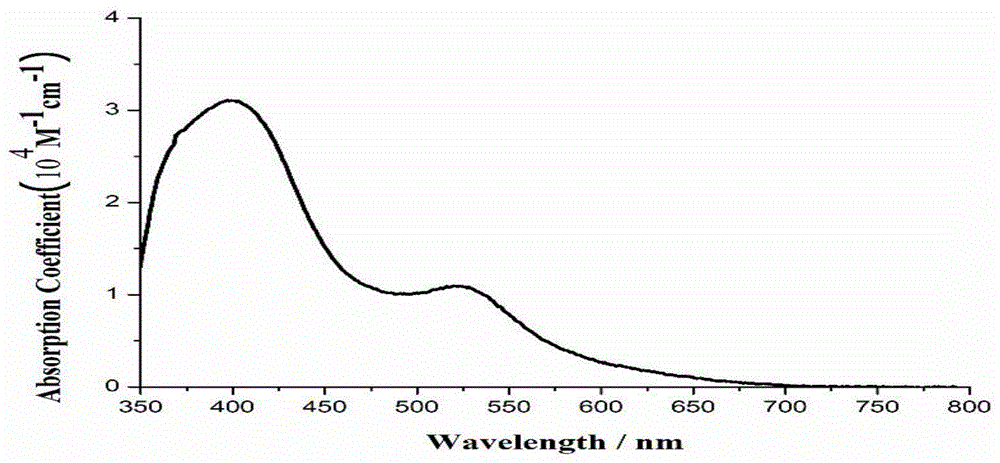
图1为实施例1染料在n,n-二甲基甲酰胺溶液中的紫外-可见吸收光谱;
图2为实施例1染料组装的染料敏化太阳能电池器件的ipce单色光电转换效率曲线;
图3为实施例1染料和明星染料n719组装的染料敏化太阳能电池器件的j-v曲线图。
具体实施方式
下面结合具体实施例,对本发明作进一步详细阐述:
实施例1
(1)将32.06mmol的5-醛基-2-噻吩硼酸、25.65mmol的3-溴-10-辛基吩噻嗪、96.18mmol的碳酸钾加入到盛有150ml的n,n-二甲基甲酰胺的三口烧瓶中,在惰性气体保护下加入2.56mmol催化量的pd(pph3)4并100℃加热,反应完毕后冷却到室温,减压旋蒸除去溶剂,然后加入200ml二氯甲烷萃取,有机相用无水硫酸钠干燥30分钟后过滤,然后减压旋蒸,剩余物用200目的硅胶、石油醚-乙酸乙酯(石油醚与乙酸乙酯体积比为10:1)进行柱层析纯化得到化合物(1),产率为75%;所得产物用1hnmr(cdcl3,400mhz)进行表征,获得以下数据:δh9.84(s,1h),7.67(d,1h),7.36-7.43(m,2h),7.24-7.26(m,2h),7.10-7.17(m,2h),6.91-6.94(m,1h),6.82-6.87(m,2h),3.84(t,2h),1.76-1.83(m,2h),1.39-1.46(m,2h),1.25-1.30(m,8h),0.86(t,3h).
(2)将11.86mmol的化合物(1)、10.67mmol的1,10-邻二氮杂菲-5,6-二酮、237.18mmol的乙酸铵加入到盛有200ml的醋酸的三口烧瓶中,在惰性气体保护下将反应液加热到120℃,反应完毕后冷却至室温,向反应液加水并过滤,然后用甲醇重结晶得到化合物(2),产率为94%;所得产物用1hnmr(dmso-d6,400mhz)进行表征,获得以下数据:δh13.85(s,1h),9.04(s,2h),8.85(t,2h),7.81-7.88(m,3h),7.55-7.61(m,3h),7.18-7.24(m,2h),7.04-7.10(m,2h),6.97(t,1h),3.91(t,2h),1.69-1.72(m,2h),1.37-1.42(m,2h),1.21-1.27(m.8h),0.83(t,3h).
(3)冰浴条件下将4.90mmol的化合物(2)加入到盛有100ml的四氢呋喃的三口烧瓶中,接着加入9.80mmol的氢化钠并搅拌2小时,然后加入7.36mmol的正溴辛烷并60℃加热,反应完毕后萃取,有机相用无水硫酸钠干燥后20分钟过滤,然后减压旋蒸,剩余物用乙醇重结晶得到化合物(3),产率为91%;所得产物用1hnmr(dmso-d6,400mhz)进行表征,获得以下数据:δh9.08(d,2h),8.89-8.92(m,1h),8.83-8.85(m,1h),7.82-7.89(m,2h),7.67-7.68(m,1h),7.57-7.63(m,3h),7.17-7.24(m,2h),7.05-7.10(m,2h),6.97(t,1h),4.90(t,2h),3.92(t,2h),1.90-1.97(m,2h),1.67-1.74(m,2h),1.34-1.42(m,2h),1.14-1.29(m,18h),0.81-0.87(m,3h),0.73-0.77(m,3h).
(4)在惰性气体保护下,将1.63mmol的二氯双(4-甲基异丙基苯基)钌(ii)和3.27mmol的化合物(3)加入到盛有50ml的n,n-二甲基甲酰胺的三口烧瓶中并加热到100℃反应4小时,接着加入3.27mmol的联吡啶双羧酸并加热到140℃反应8小时,然后加入32.66mmol的硫氰酸铵并加热到140℃反应4小时,反应停止后,加入水淬灭反应并过滤,然后先用200目的硅胶、甲醇-二氯甲烷(甲醇与二氯甲烷体积比为1:20)进行柱层析纯化,再用10ml的甲醇/石油醚进行重结晶得到化合物(a),其结构式为
以上制备过程如下所示:
实施例2
(1)将32.06mmol的5-醛基-2-噻吩硼酸、19.24mmol的3-溴-10-丁基吩噻嗪、128.24mmol的碳酸钠加入到盛有150ml的1,4-二氧六环的三口烧瓶中,在惰性气体保护下加入3.21mmol的催化量的pd(pph3)4,并95℃加热,反应完毕后冷却到室温,减压旋蒸除去溶剂,然后加入200ml的三氯甲烷萃取,有机相用无水硫酸钠干燥30分钟后过滤,然后减压旋蒸,剩余物用250目的硅胶、石油醚-乙酸乙酯(石油醚与乙酸乙酯体积比为10:1)进行柱层析纯化得到化合物(1),产率为78%;所得产物用1hnmr(cdcl3,400mhz)进行表征,获得以下数据:δh9.84(s,1h),7.67(d,1h),7.36-7.43(m,2h),7.24-7.26(m,2h),7.10-7.17(m,2h),6.91-6.94(m,1h),6.82-6.87(m,2h),3.93(t,2h),1.39-1.49(m,2h),1.25-1.29(m,2h),0.86(t,3h).
(2)将11.86mmol的化合物(1)、13.04mmol的1,10-邻二氮杂菲-5,6-二酮、118.60mmol的乙酸铵加入到盛有200ml的四氢呋喃的三口烧瓶中,在惰性气体保护下将反应液加热到100℃,反应完毕后冷却至室温,向反应液加水并过滤,然后用乙腈重结晶得到化合物(2),产率为96%;所得产物用1hnmr(dmso-d6,400mhz)进行表征,获得以下数据:δh13.85(s,1h),9.04(s,2h),8.85(t,2h),7.81-7.88(m,3h),7.55-7.61(m,3h),7.18-7.24(m,2h),7.04-7.10(m,2h),6.97(t,1h),3.93(t,2h),1.39-1.49(m,2h),1.25-1.29(m,2h),0.86(t,3h).
(3)冰浴条件下将4.90mmol的化合物(2)加入到盛有100ml的n,n-二甲基甲酰胺的三口烧瓶中,接着加入14.70mmol的氢氧化钾并搅拌3小时,然后加入12.25mmol的正氯癸烷并25℃加热,反应完毕后萃取,有机相用无水硫酸钠干燥20分钟后过滤,然后减压旋蒸,剩余物用乙醇重结晶得到化合物(3),产率为89%;所得产物用1hnmr(dmso-d6,400mhz)进行表征,获得以下数据:δh9.08(d,2h),8.89-8.92(m,1h),8.83-8.85(m,1h),7.82-7.89(m,2h),7.67-7.68(m,1h),7.57-7.63(m,3h),7.17-7.24(m,2h),7.05-7.10(m,2h),6.97(t,1h),4.64(t,2h),3.93(t,2h),1.90-1.97(m,2h),1.66-1.75(m,2h),1.14-1.29(m,18h),0.73-0.88(m,6h).
(4)在惰性气体保护下,将1.63mmol的二氯双(4-甲基异丙基苯基)钌(ii)和2.93mmol的化合物(3)加入到盛有50ml的四氢呋喃的三口烧瓶中并加热到80℃反应6小时,接着加入2.93mmol的联吡啶双羧酸并加热到120℃反应6小时,然后加入65.20mmol的硫氰酸钾并加热到160℃反应8小时,反应停止后,加入水淬灭反应并过滤,然后先用300目的硅胶、甲醇-二氯甲烷(甲醇与二氯甲烷体积比为1:20)进行柱层析纯化,再用10ml的甲醇/乙醇进行重结晶得到化合物(a),其结构式为
实施例3
(1)将32.06mmol的5-醛基-2-噻吩硼酸、32.06mmol的3-溴-10-癸基吩噻嗪、160.30mmol的磷酸钾加入到盛有150ml的四氢呋喃的三口烧瓶中,在惰性气体保护下加入1.61mmol的催化量的pd(dppf)cl2并90℃加热,反应完毕后冷却到室温,减压旋蒸除去溶剂,然后加入200ml的乙酸乙酯萃取,有机相用无水硫酸钠干燥30分钟后过滤,然后减压旋蒸,剩余物用300目的硅胶、石油醚-乙酸乙酯(石油醚与乙酸乙酯体积比为10:1)进行柱层析纯化得到化合物(1),产率为77%;所得产物用1hnmr(cdcl3,400mhz)进行表征,获得以下数据:δh9.84(s,1h),7.67(d,1h),7.36-7.43(m,2h),7.24-7.26(m,2h),7.10-7.17(m,2h),6.91-6.94(m,1h),6.82-6.87(m,2h),3.92(t,2h),1.35-1.49(m,2h),1.25-1.29(m,14h),0.87(t,3h).
(2)将11.86mmol的化合物(1)、11.86mmol的1,10-邻二氮杂菲-5,6-二酮、355.80mmol的乙酸铵加入到盛有200ml的n,n-二甲基甲酰胺的三口烧瓶中,在惰性气体保护下将反应液加热到140℃,反应完毕后冷却至室温,向反应液加水并过滤,然后用10ml乙醇/乙酸乙酯重结晶得到化合物(2),产率为91%;所得产物用1hnmr(dmso-d6,400mhz)进行表征,获得以下数据:δh13.85(s,1h),9.04(s,2h),8.85(t,2h),7.81-7.88(m,3h),7.55-7.61(m,3h),7.18-7.24(m,2h),7.04-7.10(m,2h),6.97(t,1h),3.93(t,2h),1.36-1.48(m,2h),1.21-1.29(m,14h),0.87(t,3h).
(3)冰浴条件下将4.90mmol的化合物(2)加入到盛有100ml的1,4-二氧六环的三口烧瓶中,接着加入19.60mmol的氢氧化钠并搅拌1小时,然后加入4.90mmol的正碘十二烷并80℃加热,反应完毕后萃取,有机相用无水硫酸钠干燥20分钟后过滤,然后减压旋蒸,剩余物用石油醚重结晶得到化合物(3),产率为89%;所得产物用1hnmr(dmso-d6,400mhz)进行表征,获得以下数据:δh9.08(d,2h),8.89-8.92(m,1h),8.83-8.85(m,1h),7.82-7.89(m,2h),7.67-7.68(m,1h),7.57-7.63(m,3h),7.17-7.24(m,2h),7.05-7.10(m,2h),6.97(t,1h),4.68(t,2h),3.93(t,2h),1.89-1.96(m,2h),1.64-1.76(m,2h),1.12-1.31(m,32h),0.71-0.89(m,6h).
(4)在惰性气体保护下,将1.63mmol的二氯双(4-甲基异丙基苯基)钌(ii)和3.10mmol的化合物(3)加入到盛有50ml的醋酸的三口烧瓶中并加热到120℃反应8小时,接着加入3.10mmol的联吡啶双羧酸并加热到160℃反应6小时,然后加入16.30mmol的硫氰酸钠并加热到120℃反应8小时,反应停止后,加入水淬灭反应并过滤,然后先用250目的硅胶、甲醇-二氯甲烷(甲醇与二氯甲烷体积比为1:20)进行柱层析纯化,再用甲醇进行重结晶得到化合物(a),其结构式为
实施例4
对实施例1中制备的一种高效率的联吡啶钌类染料敏化剂进行dsscs器件组装,以12微米厚度的tio2纳米晶薄膜作为光阳极,以金属铂作为对电极,将敏化好的tio2电极与pt对电极用60微米的沙林膜进行热封装,通过真空回填方式注入碘基电解质,相同条件下对明星染料n719进行组装作为对比,对组装的电池器件进行了光电转换效率性质测试,测试结果如表1、图3所示。
表1电池器件光伏性能参数
如图1所示本发所得染料具有优异的光子捕获能力,吸收光谱响应达到了近700nm;如图2所示单色-光电响应范围达到了800nm,并且在400-600nm范围内的单色光电转换效率均超过了70%。
上述为本发明的较佳实施例仅用于解释本发明,并不用于限定本发明。凡由本发明的技术方案所引伸出的显而易见的变化或变动仍处于本发明的保护范围之中。
- 还没有人留言评论。精彩留言会获得点赞!