一种UV固化透明材料的制备方法与应用与流程
本发明涉及有机高分子材料领域,具体涉及一种基于笼形聚倍半硅氧烷-碳硅烷的含硫超支化共聚物的uv固化透明材料的制备方法与应用。
背景技术:
目前传统光学透明高分子材料如聚甲基丙烯酸甲酯、聚碳酸酯等存在不耐高低温、耐老化性能差、线性膨胀系数大、易吸潮、化学稳定性差等缺点,已难以完全满足光学电子元器件等的发展需求。有机硅光学透明高分子材料具有良好耐高低温、耐候、抗老化,良好的电缘性等优点,已被广泛应用于光学透明pet离型膜、led封装、光学透镜、衍射光栅、液晶显示等领域。
现有光学透明有机硅高分子材料的固化成型基本是在加热条件下经硅氢加成反应或过氧化物自由基反应固化,或者是在有机锡催化下室温较长时间固化。这些固化方式固化时间较长、或者能耗高。紫外光固化具有固化速度快,加工工艺简单、能耗低、无污染的优点,被广泛应用到很多领域。近年来,随着人们环保意识的增强,uv固化高分子材料优势突出,展现出强大的市场竞争力。
传统uv固化高分子材料主要由四个部分组成,分别是光引发剂、基体预聚物、稀释剂以及各种助剂[陈康、李亚儒,紫外光固化交联剂研究进展,山东化工,2018,47,46-49],成本较高,残留的光引发剂会使制品性能劣化,并且固化物硬度较低,当用作光学器件保护层或者uv固化实木家具漆时,最终产品在运输过程中容易刮伤这些问题。
技术实现要素:
为解决未经改性的光学透明有机硅材料机械力学性能较差,制备中固化时间较长、或者能耗高等问题,以及传统uv固化高分子材料固化物硬度较低的问题,本发明提出了一种uv固化透明材料的制备方法,经uv固化获得硬度较高、吸水率低、热稳定性良好、透光率高的有机硅材料,材料组成简单,节约成本,材料固化后无残留的光引发剂,具有良好的抗刮伤性能。
本发明还提出一种uv固化透明材料在无溶剂uv固化实木家具漆、uv固化pet离型膜、uv固化led封装、光学透明电子器件防护层上的应用。
本发明是通过以下技术方案实现的:一种uv固化透明材料的制备方法为以下步骤:
(1)将八乙烯基笼形聚倍半硅氧烷、巯丙基烷氧基硅烷在反应溶剂a中搅拌下经uv照射反应后,减压脱溶剂和残留的原料,得到烷氧基封端的笼形聚倍半硅氧烷-碳硅烷的含硫超支化共聚物;
笼形聚倍半硅氧烷是指由硅、氧两种元素构成的无机骨架和包围在其外围的有机基团组成的分子内有机-无机杂化的化合物,将其引入聚合物体系,可以使无机相与有机相之间形成较强的化学作用,让二者之间良好相容,从而能在分子水平上对聚合物基体进行补强,并增加高分子材料的硬度和耐磨性。超支化聚碳硅烷具有良好流动性、溶解性和热稳定性等。
作为优选,八乙烯基聚倍半硅氧烷与巯丙基硅氧烷的使用量中乙烯基与巯丙基的摩尔比为1∶1~1∶1.2。巯基硅氧烷与八乙烯基聚倍半硅氧烷反应,它不会交联固化,只是让它充分反应。此时,乙烯基与巯丙基摩尔比1∶1~1∶1.2,是为了保证乙烯基完全反应,保证所得超支化聚合物的结构可控性和规整性。
作为优选,巯丙基烷氧基硅烷选自巯丙基三甲氧基硅烷、巯丙基三乙氧基硅烷、巯丙基甲基二甲氧基硅烷、巯丙基甲基二乙氧基硅烷中的一种或几种。
作为优选,反应溶剂a选自四氢呋喃、乙醚、甲苯、二甲苯、石油醚中的一种或几种。使用量为使溶质溶解的量,优选为八乙烯基聚倍半硅氧烷和巯丙基硅氧烷总质量的1-2倍。
uv照射反应条件为:光源功率100w-1000w,主峰波长365nm或405nm,距离光源20cm,照射反应0.5~6h。
作为优选,通过80~120℃/130mmhg减压脱溶剂和残留的原料。
反应式为(以合成24个甲氧基封端的笼形聚倍半硅氧烷-碳硅烷的含硫超支化共聚物为例):
(2)将步骤(1)产物在无水无氧条件下,经格氏反应获得笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物;反应式为(以合成含24个烯丙基的液体笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物为例):
具体制备方法为:
(a)在无水无氧条件下,将60gmg屑(2.500mol),一到两粒碘置于洁净的三口瓶中,随后加入400~3000ml溶剂b,将混合体系用冰浴降温到-5~20℃后,缓慢滴加50~600ml溶剂b和252.0g烯丙基溴(2.100mol,过量5%)的混合溶液,滴加完后,-5~20℃反应1~24h,获得烯丙基溴化镁格氏试剂的溶剂b溶液。
溶剂b选自四氢呋喃、乙醚中的一种或两的混合物。
(b)将20ml溶剂b溶解干燥的烷氧基封端的笼形聚倍半硅氧烷-碳硅烷的含硫超支化共聚物,将其缓慢滴加到烯丙基溴格氏试剂b溶液中,加完后,将反应混合物维持-5~40℃反应8~24h。将反应混合物加入到-5~40℃的50~500ml饱和nh4cl水溶液中(饱和nh4cl水溶液用量以搅拌混合后无气泡冒出为宜),用布氏漏斗抽滤。水相用30~300ml溶剂b萃取三次,合并的有机相用30~300ml去离子水洗涤三次后,用50~200ml饱和nacl水溶液洗涤一次,接着用20~50g无水mgso4干燥后,80~120℃/130mmhg减压除去溶剂和未反应的原料,获得粗产品。然后将粗产品倒入50~500ml乙醇中,粗产物沉淀、40~80℃/130mmhg干燥12~24h,得到笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物。
(3)将笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物与含巯基的聚硅氧烷混合、脱出气泡后经uv固化10~120s,得到uv固化透明材料。
作为优选,笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物与含巯基的有机硅聚合物中巯基与烯丙基摩尔比为0.6∶1~1.3∶1。作为优选,笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物和含巯基的有机硅聚合物按照烯丙基和巯基摩尔比为0.8∶1~1.2∶1混合均匀。此时体系会在uv辐射下发生交联反应。
作为优选,在130mmhg/25~40℃减压脱气泡5~30min。
作为优选,uv固化光源功率500-1000w,主峰波长365nm或405nm,距离光源20cm。
含巯基的有机硅聚合物的制备方法为:含巯基的烷氧基硅烷与其他烷氧基硅氧烷、封端剂六甲基二硅氧烷在有机溶剂c中,在酸性催化剂催化下共水解-缩合后,水洗至中性,再减压脱除有机溶剂c、残留原料和低分子产物后得到含巯基聚硅氧烷,其中,每个分子中至少含有三个巯丙基官能团。
所述含巯基的烷氧基硅烷选自巯丙基甲基二甲氧基硅烷、巯丙基甲基二乙氧基硅烷、巯丙基三甲氧基硅烷、巯丙基三乙氧基硅烷中的一种或几种。
所述其他烷氧基硅烷选自二甲基二甲氧基硅烷、二甲基二乙氧基硅烷、甲基苯基二甲氧基硅烷、甲基苯基二乙氧基硅烷、二苯基二甲氧基硅烷、二苯基二乙氧基硅烷、甲基三甲氧基硅烷、甲基三乙氧基硅烷、苯基三甲氧基硅烷、苯基三乙氧基硅烷、正硅酸甲酯、正硅酸乙酯中的一种或几种;作为优选,其他烷氧基硅烷选自二甲基二乙氧基硅烷、甲基苯基二甲氧基硅烷、甲基苯基二乙氧基硅烷、甲基三甲氧基硅烷、苯基三甲氧基硅烷、正硅酸乙酯中的一种或几种。
含巯基的硅氧烷与其他烷氧基硅烷摩尔比为0.05~0.65∶1,所有有机基团与硅原子摩尔比为1.3~2.0∶1,作为优选,含巯基的硅氧烷与其他烷氧基硅烷摩尔比为0.1~0.45∶1,所有有机基团与硅原子摩尔比为1.4~1.8∶1。
所述有机溶剂c选自甲苯、二甲苯、石油醚、四氢呋喃、醋酸丁酯中的一种或几种;其用量为原料总质量的0.5~4倍。作为优选,有机溶剂选自甲苯、二甲苯、石油醚中的一种或几种,其用量为原料总质量的0.8~2倍。
所述酸性催化剂选自盐酸、硫酸、醋酸、三氟甲磺酸、对甲苯磺酸中的一种或几种,催化剂用量为原料总质量的0.05~5wt%,作为优选,酸性催化剂用量为原料总质量的0.1~3wt%。
水解-缩合反应中水的用量为原料中所有烷氧基摩尔数的1~2.5倍,作为优选,水的用量为原料中所有烷氧基摩尔数的1~1.8倍。
共水解-缩合反应温度为30~80℃,反应时间为0.5~48h。作为优选,共水解-缩合反应温度为40~70℃,反应时间为2~18h。
uv固化透明材料制备方法中,光固化条件为:uv固化光源功率500-1000w,主峰波长365nm或405nm,距离光源20cm,优选为zb-t150-uv1000型微波无线电uv光固化机。
本发明合成笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物,并将其与含巯基的有机硅聚合物经巯基-乙烯基(thiol-ene)点击反应,经uv固化获得硬度较高、吸水率低、热稳定性良好、透光率高的有机硅材料。材料制备过程中不需使用是光引发剂和稀释剂和各种助剂,材料组成简单,节约成本,便于工业化,材料固化后具有良好的抗刮伤性能。
制备得到的uv固化透明材料在400~800nm光波范围内透光率到达90%以上,铅笔硬度在b~7h范围内可控,吸水率0.02wt%~0.25wt%,初始热分解260℃~360℃。所得透明有机硅涂层材料可望用于无溶剂uv固化实木家具漆、uv固化pet离型膜、uv固化led封装、光学透明电子器件防护层。
与现有技术相比,本发明的有益效果是:
(1)克服了现有有机硅光学透明材料制备过程中固化时间较长、或者能耗高等问题;无需加入光引发剂、稀释剂和其他助剂,克服了现有uv固化高分子材料组成较复杂(主要由四个部分组成,分别是光引发剂、基体预聚物、稀释剂以及各种助剂),成本较高,残留光引发剂影响产品质量等问题;
(2)经uv固化获得硬度较高、吸水率低、热稳定性良好、透光率高的有机硅材料,具有良好的抗刮伤性能。
附图说明
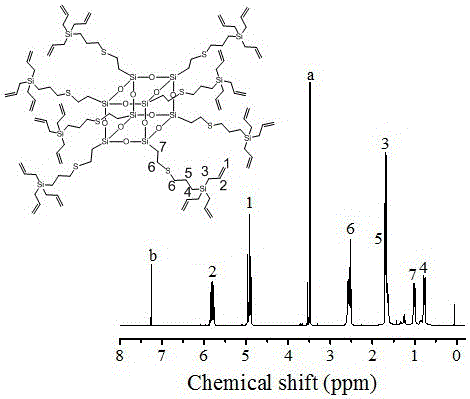
图1为实施例1中含24个烯丙基的笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物的1hnmr谱图;
图2为实施例1中含24个烯丙基的笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物poss-vi24、巯基树脂、固化膜的ir谱图;
图3为实施例1中含24个烯丙基的笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物与0.004%mol/g巯基含量,不同r/si的巯基聚硅氧烷固化物照片;
图4为实施例1中含24个烯丙基的笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物与0.004%mol/g巯基含量,不同巯基与烯丙基摩尔比的固化物照片;
图5为表1中12的固化物透光率曲线。
具体实施方式
下面通过实施例对本发明作进一步详细说明,实施例中所用原料均可市购或采用常规方法制备。
核磁共振:以氘代氯仿(cdcl3)作溶剂,用德国brucker公司bruckeradvance-400nmr核磁共振仪室温下测氢谱(1h-nmr)。
傅立叶红外(ft-ir):使用美国nicolet公司nicolet700型傅里叶变换红外光谱仪(配有atr附件),测试时将样品直接放在红外光谱仪atr附件中,测试在4000~700cm-1范围内的傅立叶红外谱图。
透光率测试:美国thermofisher公司的evolution300型紫外可见分光光度计测试聚合物的透光率,测试波长范围为400~800nm,样品厚度10mm;
铅笔硬度:按gb/t6739-2006《色漆和清漆铅笔法测定漆膜硬度》进行测定。
热重分析(tga):用德国耐驰仪器制造有限公司tg209热重分析仪进行热重分析,氮气保护下,升温速率10℃/min,温度范围为室温~800℃。
水接触角:il4200型接触角测量仪,德国kruss公司,将吸有去离子水的针管由微型注射器滴水2微升到待测样品表面,测定蒸馏水在空气中与固体涂膜的接触角,内切法测定其值。平行测定5次取平均值。
吸水率:将涂膜剪成一定形状的方块,室温下在去离子水中浸泡24h,用滤纸吸干涂膜表面的水,按式计算涂膜的吸水率。
其中b代表吸水率(%);m1表示浸泡前涂膜的质量;m2表示浸泡之后使用滤纸吸干涂膜表面液体后的质量。
实施例1
(1)将1.0g八乙烯基聚倍半硅氧烷(poss-vi8,1.58mmol),2.48g(3-巯基丙基)三甲氧基硅烷(12.64mmol)和30mlthf加入装有搅拌棒的50ml单颈圆底烧瓶中。将反应混合物经主峰波长405nm500w的uv光照射反应3h后,通过120℃/130mmhg减压脱溶剂和残留的原料,获得24个甲氧基封端的笼形聚倍半硅氧烷-碳硅烷的含硫超支化共聚物。
(2)在无水无氧条件下,将60gmg屑(2.500mol),两粒碘置于洁净的三口瓶中,随后加入1000ml乙醚,将混合体系用冰浴降温到0~5℃后,缓慢滴加200ml乙醚和252.0g烯丙基溴(2.100mol)的混合溶液。滴加完后,0~5℃反应2h,获得烯丙基溴化镁格氏试剂的乙醚溶液。
将20ml干燥乙醚和所得的24个甲氧基封端的笼形聚倍半硅氧烷-碳硅烷的含硫超支化共聚物混合物缓慢滴加到烯丙基溴格氏试剂中,加完后,将反应混合物室温搅拌过夜。将反应混合物加入到冷却的100ml饱和nh4cl水溶液中,抽滤。水层用乙醚50ml萃取三次,合并的有机层用去离子水50ml洗涤三次后,用50ml饱和nacl水溶液洗涤一次,接着用20g无水mgso4干燥后,除去大部分挥发物,然后通过将浓缩溶液倒入100ml甲醇中使粗产物沉淀为油状物。真空干燥纯产物,得到6g可流动的粘稠液体,即为含24个烯丙基的笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物,1hnmr谱图如图1所示。
1hnmr(400mhz,cdcl3,ppm):δ=0.63-0.92(-sich2ch2-),0.93-0.1.14(-sich2-),1.18-1.39(-ch2ch2ch2-),1.51-1.84(-sich2ch=ch2),2.38-2.80(-ch2sch2-),4.68-5.01(-ch2ch=ch2),5.60-5.95(-ch2ch=ch2).a为残存的si-o-ch3,b为cdcl3溶剂峰。
按氢谱计算双键含量为:7.41mmol/g。
(3)、在50℃和机械搅拌下,向78.54g3-巯丙基三甲氧基硅烷、48.75g二甲二乙氧基硅烷、47.98g甲基三甲氧硅烷、1.62g六甲基二硅氧烷(按照巯基含量0.004mol/g,有机基团/硅原子为1.3的配比)和175g甲苯的混合物中滴加56.2g去离子水和3.5g36.5%的浓盐酸的混合物,0.5h滴完后继续在50℃下反应18h,然后水洗至中性,并在130mmhg/170℃下减压脱出溶剂、残留原料和低分子产物,获得无色透明的含巯基聚硅氧烷70.5g。
取步骤(2)中液体笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物3.0g、巯基聚硅氧烷5.76g,混合均匀后,经130mmhg/30℃真空脱泡20min后,经主峰波长405nmuv固化30s成膜,得到uv固化透明材料1。
uv固化透明材料1透光率90.5%,铅笔硬度3h,热分解温度330℃。
如图2所示,固化膜在1627cm-1碳碳双键的伸缩振动特征峰几乎消失;在2550cm-1巯基的特征峰消失。可以看出固化体系中巯基和烯丙基反应良好,可以证明固化体系制备成功。
实施例2
(1)将1.0g八乙烯基聚倍半硅氧烷(poss-vi8,1.58mmol),3.014g(3-巯基丙基)三乙氧基硅烷(12.64mmol)和100ml乙醚加入装有搅拌棒的250ml单颈圆底烧瓶中。将反应混合物经100w的主峰波长365nmuv光照射反应6h后,通过80℃/130mmhg减压脱溶剂和残留的原料,获得24个乙氧基封端的笼形聚倍半硅氧烷-碳硅烷的含硫超支化共聚物。
(2)在无水无氧条件下,将60gmg屑(2.500mol),两粒碘置于洁净的三口瓶中,随后加入400ml四氢呋喃,将混合体系用冰浴降温到0~5℃后,缓慢滴加50ml四氢呋喃和252.0g烯丙基溴(2.100mol)的混合溶液。滴加完后,-5~5℃反应24h,获得烯丙基溴化镁格氏试剂的四氢呋喃溶液。
将20ml干燥乙醚和24个乙氧基封端的笼形聚倍半硅氧烷-碳硅烷的含硫超支化共聚物混合物缓慢滴加到烯丙基溴格氏试剂中,加完后,将反应混合物室温搅拌过夜。将反应混合物加入到冷却的200ml饱和nh4cl水溶液中,抽滤。水层用四氢呋喃300ml萃取三次,合并的有机层用去离子水300ml洗涤三次后,用200ml饱和nacl水溶液洗涤一次,接着用30g无水mgso4干燥后,除去大部分挥发物,然后通过将浓缩溶液倒入500ml乙醇中使粗产物沉淀为油状物。真空干燥纯产物,得到6.8g可流动的粘稠液体、即为含24个烯丙基的笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物。
(3)在40℃和机械搅拌下,向78.54g3-巯丙基三甲氧基硅烷、64.34g二甲二乙氧基硅烷、32.83g甲基三甲氧硅烷、1.62g六甲基二硅氧烷(按照巯基含量0.004mol/g,有机基团/硅原子为1.4的配比)和351.4g二甲苯的混合物中滴加109.2g去离子水和8.785g对甲苯磺酸的混合物,0.5h滴完后继续在40℃下反应24h,然后水洗至中性,并在130mmhg/170℃下减压脱出溶剂、残留原料和低分子产物,获得无色透明的含巯基聚硅氧烷75.8g。
按照如下表中的配比,分别取巯基聚硅氧烷和1中笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物,混合均匀后,经130mmhg/30℃真空脱泡20min后,uv固化透明材料2-1到2-17。
uv固化透明材料2-1到2-17经主峰波长365nmuv固化后,其结果如表1所示。
表1不同巯基与烯丙基摩尔比的巯基聚硅氧烷与笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物经uv固化的材料性能列表
表1中12的固化物透光率曲线如图5所示,说明固化物在可见光范围内(400-800nm)有非常高的透光率。
实施例3
(1)将1.0g八乙烯基聚倍半硅氧烷(poss-vi8,1.58mmol),2.28g(3-巯基丙基)甲基二甲氧基硅烷(12.64mmol)、50ml四氢呋喃和50ml乙醚加入装有搅拌棒的250ml单颈圆底烧瓶中。将反应混合物经1000w的主峰波长365nmuv光照射反应0.5h后,通过80℃/130mmhg减压脱溶剂和残留的原料,获得8个甲基二甲氧基封端的笼形聚倍半硅氧烷-碳硅烷的含硫超支化共聚物。
(2)在无水无氧条件下,将60gmg屑(2.500mol),两粒碘置于洁净的三口瓶中,随后加入1000ml四氢呋喃、1000ml二甲苯和1000ml乙醚,将混合体系用冰浴降温到20~30℃后,缓慢滴加100ml四氢呋喃、200ml二甲苯、200ml乙醚和252.0g烯丙基溴(2.100mol)的混合溶液。滴加完后,20~30℃反应12h,获得烯丙基溴化镁格氏试剂的溶液。
将40ml干燥乙醚和8个甲基二甲氧基封端的笼形聚倍半硅氧烷-碳硅烷的含硫超支化共聚物混合物缓慢滴加到烯丙基溴格氏试剂中,加完后,将反应混合物室温搅拌过夜。将反应混合物加入到冷却的500ml饱和nh4cl水溶液中,抽滤。水层用四氢呋喃100ml、乙醚200ml萃取三次,合并的有机层用去离子水100ml洗涤三次后,用100ml饱和nacl水溶液洗涤一次,接着用40g无水mgso4干燥后,除去大部分挥发物,然后通过将浓缩溶液倒入200ml甲醇和100ml异丙醇中使粗产物沉淀为油状物。真空干燥纯产物,得到7.0g可流动的粘稠液、即为8个甲基二烯丙基封端的体笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物。
(3)在30℃和机械搅拌下,向78.54g3-巯丙基三甲氧基硅烷、79.63g二甲二乙氧基硅烷、17.97g甲基三甲氧硅烷、1.62g六甲基二硅氧烷(按照巯基含量0.004mol/g,有机基团/硅原子为1.5的配比)和744.6g四氢呋喃的混合物中滴加132.48g去离子水和8.785g三氟甲磺酸的混合物,0.5h滴完后继续在30℃下反应48h,然后水洗至中性,并在130mmhg/170℃下减压脱出溶剂、残留原料和低分子产物,获得无色透明的含巯基聚硅氧烷68.5g。
取巯基聚硅氧烷6.669g、8个甲基二烯丙基封端的体笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物3.0g,混合均匀后,经130mmhg/30℃真空脱泡20min后,经主峰波长405nmuv固化30s成膜,所得uv固化透明材料3。
uv固化透明材料3透光率90.8%,铅笔硬度5h,热分解温度354.5℃。
实施例4
(1)将1.0g八乙烯基聚倍半硅氧烷(poss-vi8,1.58mmol),2.456g(3-巯基丙基)甲基二乙氧基硅烷(12.64mmol)、50ml四氢呋喃和50ml乙醚加入装有搅拌棒的250ml单颈圆底烧瓶中。将反应混合物经200w的主峰波长405nmuv光照射反应4h后,通过80℃/130mmhg减压脱溶剂和残留的原料,获得8个甲基二乙氧基甲氧基封端的笼形聚倍半硅氧烷-碳硅烷的含硫超支化共聚物。
(2)在无水无氧条件下,将60gmg屑(2.500mol),两粒碘置于洁净的三口瓶中,随后加入1000ml四氢呋喃和1000ml甲苯,将混合体系用冰浴降温到20-30℃后,缓慢滴加300ml四氢呋喃、200ml甲苯和252.0g烯丙基溴(2.100mol)的混合溶液。滴加完后,30-40℃反应6h,获得烯丙基溴化镁格氏试剂的溶液。
将40ml干燥乙醚和8个甲基二乙氧基甲氧基封端的笼形聚倍半硅氧烷-碳硅烷的含硫超支化共聚物混合物缓慢滴加到烯丙基溴格氏试剂中,加完后,将反应混合物室温搅拌过夜。将反应混合物加入到冷却的200ml饱和nh4cl水溶液中,抽滤。水层用四氢呋喃100ml、甲苯200ml萃取三次,合并的有机层用去离子水100ml洗涤三次后,用100ml饱和nacl水溶液洗涤一次,接着用40g无水mgso4干燥后,除去大部分挥发物,然后通过将浓缩溶液倒入200ml甲醇和100ml异丙醇中使粗产物沉淀为油状物。真空干燥纯产物,得到6.5g可流动的粘稠液体、即为8个甲基二烯丙基封端的体笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物。
(3)在70℃和机械搅拌下,向78.54g3-巯丙基三甲氧基硅烷、36.6g巯丙基甲基二甲氧基硅烷、79.63g二甲二乙氧基硅烷、17.97g甲基三甲氧硅烷、正硅酸乙酯10.4g和1.62g六甲基二硅氧烷(按照巯基含量0.0065mol/g,有机基团/硅原子为2.0的配比)和224.2g醋酸丁酯的混合物中滴加132.48g去离子水和1.1157g浓硫酸的混合物,0.5h滴完后继续在70℃下反应4h,然后水洗至中性,并在130mmhg/170℃下减压脱出溶剂、残留原料和低分子产物,获得无色透明的含巯基聚硅氧烷89.2g。
取巯基聚硅氧烷4.104g、8个甲基二烯丙基封端的体笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物3.0g,混合均匀后,经130mmhg/30℃真空脱泡30min后,经主峰波长365nmuv固化30s成膜,所得uv固化透明材料4。
uv固化透明材料4透光率91.5%,铅笔硬度3h,热分解温度334.5℃。
实施例5
步骤(1)与(2)同实施例1。
(3)在70℃和机械搅拌下,向39.27g3-巯丙基三甲氧基硅烷、18.3g巯丙基甲基二甲氧基硅烷、79.63g二甲二乙氧基硅烷、39.6g甲基苯基二甲氧基硅烷、17.97g甲基三甲氧硅烷、19.8g苯基三甲氧基硅烷、正硅酸乙酯10.4g和1.62g六甲基二硅氧烷(按照巯基含量0.002mol/g,有机基团/硅原子为1.3的配比)和350g石油醚的混合物中滴加155.5g去离子水和3.5g醋酸的混合物,0.5h滴完后继续在75℃下反应12h,然后水洗至中性,并在130mmhg/170℃下减压脱出溶剂、残留原料和低分子产物,获得无色透明的含巯基聚硅氧烷98.5g。
取巯基聚硅氧烷13.338g、实施例1中含24个烯丙基的笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物3.0g,混合均匀后,经130mmhg/30℃真空脱泡5min后,经主峰波长405nmuv固化30s成膜,所得uv固化透明材料5。
uv固化透明材料5透光率92.5%,铅笔硬度3h,热分解温度338.5℃。
实施例6
步骤(1)与(2)同实施例1。
(3)在80℃和机械搅拌下,向42.56g3-巯丙基三乙氧基硅烷、22.8g巯丙基甲基二乙氧基硅烷、79.63g二甲二乙氧基硅烷、39.6g二苯基二乙氧基硅烷、17.97g甲基三乙氧硅烷、19.8g苯基三乙氧基硅烷、正硅酸甲酯11.8g和1.62g六甲基二硅氧烷(按照巯基含量0.3mol/g,有机基团/硅原子为1.5的配比)和350g石油醚的混合物中滴加155.5g去离子水和3.5g36.5%浓盐酸的混合物,0.5h滴完后继续在80℃下反应0.5h,然后水洗至中性,并在130mmhg/170℃下减压脱出溶剂、残留原料和低分子产物,获得无色透明的含巯基聚硅氧烷98.5g。
取巯基聚硅氧烷8.892g、实施例1中含24个烯丙基的笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物3.0g,混合均匀后,经130mmhg/30℃真空脱泡5min后,经主峰波长405nmuv固化30s成膜,所得uv固化透明材料6。
uv固化透明材料6透光率91.8%,铅笔硬度4h,热分解温度346.0℃。
实施例7
步骤(1)与(2)同实施例1。
(3)在80℃和机械搅拌下,向52.85g3-巯丙基三乙氧基硅烷、32.8g巯丙基甲基二乙氧基硅烷、79.63g二甲二乙氧基硅烷、39.6g二苯基二乙氧基硅烷、17.97g甲基三乙氧硅烷、19.8g苯基三乙氧基硅烷、正硅酸甲酯11.8g和1.62g六甲基二硅氧烷(按照巯基含量0.45mol/g,有机基团/硅原子为1.5的配比)、100g甲苯和150g二甲苯的混合物中滴加155.5g去离子水和3.5g36.5%浓盐酸的混合物,0.5h滴完后继续在80℃下反应0.5h,然后水洗至中性,并在130mmhg/170℃下减压脱出溶剂、残留原料和低分子产物,获得无色透明的含巯基聚硅氧烷98.5g。
取巯基聚硅氧烷5.928g、实施例1中含24个烯丙基的笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物3.0g,混合均匀后,经130mmhg/30℃真空脱泡5min后,经主峰波长365nmuv固化30s成膜,所得uv固化透明材料7。
uv固化透明材料7透光率90.4%,铅笔硬度6h,热分解温度356.0℃。
测试例
实施例1中含24个烯丙基的笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物与0.004%mol/g巯基含量,不同r/si的巯基聚硅氧烷固化物照片如图3所示,含24个烯丙基的笼形聚倍半硅氧烷-碳硅烷的烯丙基封端超支化共聚物与0.004%mol/g巯基含量,不同巯基与烯丙基摩尔比的固化物照片如图4所示。由此可见,本申请的uv固化透明材料透明度好。
对比例
采用实施例2的制备过程,步骤(3)中的固化时间分别采用5秒。
说明固化时间太短会影响uv固化透明材料的铅笔硬度。
实施例制备得到的uv固化透明材料在400~800nm光波范围内透光率到达90%以上,铅笔硬度在b~7h范围内可控,吸水率0.02wt%~0.25wt%,初始热分解260℃~360℃。所得透明有机硅涂层材料具有较好的抗刮伤性能,可望用于无溶剂uv固化实木家具漆、uv固化pet离型膜、uv固化led封装、光学透明电子器件防护层。
- 还没有人留言评论。精彩留言会获得点赞!