一种可见光促进β-胺基硒醚的合成方法与流程
本发明涉及有机合成化学技术领域,具体为一种可见光促进β-胺基硒醚的合成方法。
背景技术:
含氮化合物是非常重要的有机化合物,而且大多数具有较强的生物活性,其大量存在于天然产物中,尤其是在含氮杂环的抗肿瘤药物的合成中具有极其重要的意义(ricci,a.,aminogroupchemistry:fromsynthesistothelifesciences,1sted.,wiley-vch,weinheim,2007)。另一方面,硒元素是人体的必需微量元素,此外,有机硒化合物在有机合成中有着广泛的应用,如可以用作还原剂、氧化剂、催化剂、亲核试剂和亲电试剂等((a)klayman,d.l.;günther,w.h.h.organicseleniumcompounds:theirchemistryandbiology;johnwiley&sons,newyork,1973.;(b)nogueira,c.w.;zeni,g.;rocha,j.b.t.chem.rev.2004,104,6255-6286.)。因此,发展新颖高效、环境友好的合成含氮含硒化合物方法是目前有机合成领域研究的热点之一。
合成β-胺基硒醚化合物主要通过烯烃的硒胺化反应。然而目前所报道的该类方法存在一定的缺点,比如需要使用酸、氧化剂和过渡金属等添加剂;使用过量的有机硒源;以及使用预先制备有机硒试剂作为反应前体((a)sun,k.;wang,x.;lv,y.;li,g.;jiao,h.;dai,c.;li,y.;zhang,c.;liu,l.,chem.commun.2016,52(54),8471-8474;(b)tang,e.;wang,w.;zhao,y.;zhang,m.;dai,x.,org.lett.2016,18(2),176-179.;(c)toshimitsu,a.;aoai,t.;owada,h.;uemura,s.;okano,m.,j.org.chem.1981,46(23),4727-4733.;(d)tiecco,m.;testaferri,l.;santi,c.;tomassini,c.;marini,f.;bagnoli,l.;temperini,a.,angew.chem.int.ed.2003,42(27),3131-3133.)。
可见光是一种清洁无污染的能源,近年来可见光促进的有机合成反应得到了极大的发展。然而文献调研表明,基于可见光促进β-胺基硒醚合成方法处于起步阶段,并且在有限的文献报道中通常需要加入一定量的光催化剂。在此,我们报道一种不使用任何添加剂条件下,可见光促进烯烃硒胺化反应合成β-胺基硒醚化合物新合成方法。
技术实现要素:
本发明的目的在于提供一种具有反应条件温和,产率高,官能团兼容性好及原子经济性等优势的可见光促进β-胺基硒醚的合成方法,以解决上述背景技术中提出的问题。
为实现上述目的,本发明提供如下技术方案:一种可见光促进β-胺基硒醚的合成方法,包括以下步骤:
a、在反应器中依次加入化合物1,2,3,并通入惰性气体;
b、在一定温度条件下和光源照射下进行搅拌反应;
c、反应结束后减压蒸除溶剂得粗产物;
d、经柱层析提纯得到β-胺基硒醚4。
优选的,所述根据步骤a反应器是指schlenk管,反应过程中使用的惰性气体为氮气或者氩气,通过化学反应制备得到了β-胺基硒醚化合物,其反应方程式如下:
式中化合物1是可以为苯乙烯,各被取代的苯乙烯、或表示各任选被取代的具有5-10个环原子的单或双环杂芳基乙烯,化合物2可以为糖精及其衍生物,双苯磺酰亚胺,l-(+)-樟脑内磺酰胺,咪唑,1,2,4-三氮唑,苯并吡唑等,所述化合物3可以二芳基二硒醚,二烷基二硒醚。
优选的,所述根据步骤a所述化合物的摩尔比为1:1:0.5。
优选的,所述根据步骤b在室温条件下进行反应,室温反应下采用23瓦的紧凑型荧光灯发出的白色光线照射。
优选的,所述根据步骤c溶剂是乙腈,二氯甲烷,二甲基甲酰胺,乙酸乙酯中的一种。
优选的,所述根据步骤d柱层析提纯所用的洗脱液为石油醚与乙酸乙酯的混合溶剂,所述石油醚:乙酸乙酯的体积比为(0.5~50):1的混合溶剂。
与现有技术相比,本发明的有益效果是:
本发明制备方法以烯烃,含氮化合物,二硒醚为原料,以乙腈,二氯甲烷、二甲基甲酰胺,乙酸乙酯中的一种为溶剂,反应温度为室温,白色荧光的光源照射下,高效合成出β-胺基硒醚化合物,该方法与传统的合成方法相比,具有反应条件温和,产率高,官能团兼容性好及原子经济性等优势。
附图说明
图1为本发明10:1洗脱剂所得产物的氢谱图和碳谱图;
图2为本发明7:1洗脱剂所得产物的氢谱图和碳谱图;
图3为本发明9:1洗脱剂所得产物的氢谱图和碳谱图;
图4为本发明12:1洗脱剂所得产物的氢谱图和碳谱图;
图5为本发明10:1洗脱剂所得产物的氢谱图和碳谱图;
图6为本发明11:1洗脱剂所得产物的氢谱图和碳谱图;
图7为本发明9:1洗脱剂所得产物的氢谱图和碳谱图;
图8为本发明10:1洗脱剂所得产物的氢谱图和碳谱图;
图9为本发明7:1洗脱剂所得产物的氢谱图和碳谱图;
图10为本发明10:1洗脱剂所得产物的氢谱图和碳谱图;
图11为本发明7:1洗脱剂所得产物的氢谱图和碳谱图;
图12为本发明9:1洗脱剂所得产物的氢谱图和碳谱图;
图13为本发明12:1洗脱剂所得产物的氢谱图和碳谱图;
图14为本发明10:1洗脱剂所得产物的氢谱图和碳谱图;
图15为本发明11:1洗脱剂所得产物的氢谱图和碳谱图;
图16为本发明9:1洗脱剂所得产物的氢谱图和碳谱图;
图17为本发明10:1洗脱剂所得产物的氢谱图和碳谱图;
图18为本发明7:1洗脱剂所得产物的氢谱图和碳谱图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例,基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围,
请参阅图1-18,本发明提供一种技术方案:一种以烯烃1原料,反应中所用氮源2为糖精及其衍生物,双苯磺酰亚胺,l-(+)-樟脑内磺酰胺,咪唑,1,2,4-三氮唑、苯并吡唑等等,硒源3为二芳基二硒醚,二烷基二硒醚,室温下,向装有2.0~3.0毫升乙腈,二氯甲烷,二甲基甲酰胺,乙酸乙酯中的一种的25毫升schlenk,管中装入烯烃10.5毫摩尔,氮源20.5毫摩尔和二硒醚30.25毫摩尔,加毕,通入惰性气体,将23瓦白色紧凑型荧光灯放在距离反应管3厘米的地方,室温下反应20小时,反应完成后,将反应液通过旋转蒸发器除去溶剂,残余物用硅胶柱进行纯化,硅胶规格为200目~300目,柱层析提纯所用的洗脱液为石油醚与乙酸乙酯的混合溶剂,石油醚:乙酸乙酯的体积比为(0.5~50):1的混合溶剂,得到目标产物β-胺基硒醚,产率视不同反应在65%~92%之间。
实施例1
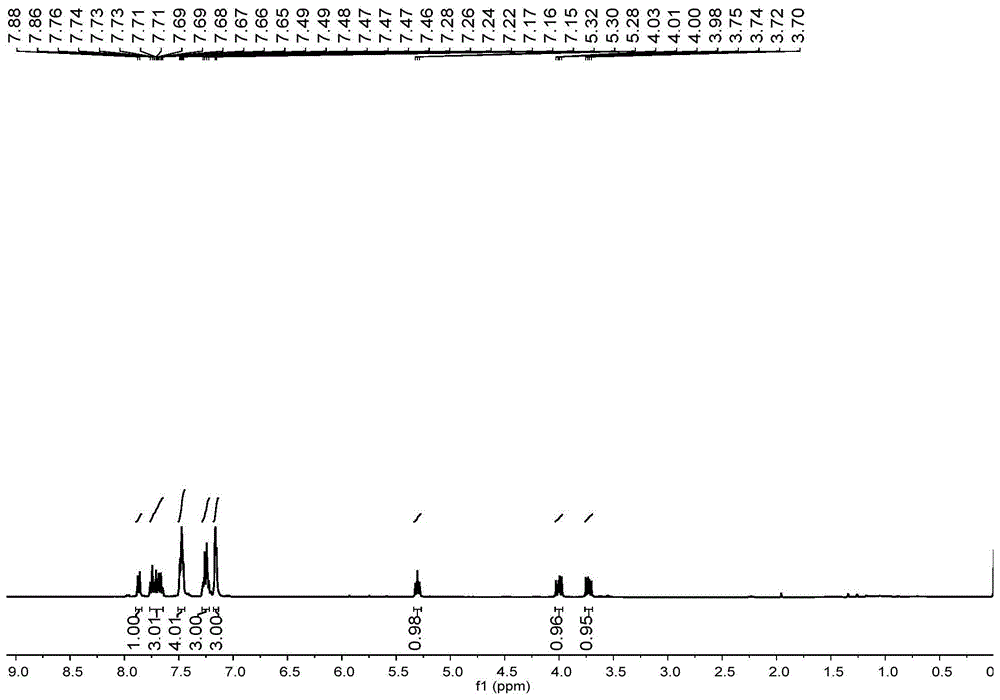
室温下,向装有磁力搅拌子的25毫升schlenk(施兰克)管中装入过苯乙烯(0.5毫摩尔),糖精(0.5毫摩尔),二苯基二硒醚(0.25毫摩尔),乙腈(2毫升),加毕,充入n2保护,将23瓦白色紧凑型荧光灯放在距离反应管3厘米的地方,室温下反应20小时,反应完成后,将有机相通过旋转蒸发器除去溶剂,残余物用硅胶柱进行纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯=10:1),得到白色固体194毫克,产率为92%,
所得产物核磁图谱数据为:1hnmr(400mhz,cdcl3):δ/ppm=7.87(d,j=7.0hz,1h),7.70(ddt,j=14.8,7.3,6.6hz,3h),7.50–7.45(m,4h),7.29–7.22(m,3h),7.17–7.14(m,3h),5.30(t,j=8.1hz,1h),4.00(dd,j=12.9,8.1hz,1h),3.73(dd,j=12.9,8.1hz,1h).13cnmr(100mhz,cdcl3):δ/ppm=157.7,136.3,135.6,133.7,133.2,132.7,128.1,128.0,127.7,127.5,127.4,126.6,126.0,124.1,119.7,56.8,28.0。
实施例2
室温下,向装有磁力搅拌子的25毫升schlenk(施兰克)管中装入过4-甲氧基苯乙烯(0.5毫摩尔),糖精(0.5毫摩尔),二苯基二硒醚(0.25毫摩尔),乙腈(2毫升),加毕,充入n2保护,将23瓦白色紧凑型荧光灯放在距离反应管3厘米的地方,室温下反应20小时,反应完成后,将有机相通过旋转蒸发器除去溶剂,残余物用硅胶柱进行纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯=7:1),得到白色固体210毫克,产率为89%,
所得产物核磁图谱数据为:1hnmr(400mhz,cdcl3):δ/ppm=7.86(dd,j=6.8,1.2hz,1h),7.77–7.64(m,3h),7.49–7.39(m,4h),7.18–7.14(m,3h),6.78(d,j=8.8hz,2h),5.28(t,j=8.1hz,1h),3.95(dd,j=12.8,8.1hz,1h),3.74(dd,j=12.8,8.1hz,1h),3.69(s,3h).13cnmr(100mhz,cdcl3):δ/ppm=158.7,157.6,136.4,133.7,133.2,132.6,129.0,128.1,128.0,127.5,126.5,126.1,124.1,119.7,112.8,56.3,54.2,28.1。
实施例3
室温下,向装有磁力搅拌子的25毫升schlenk(施兰克)管中装入过2-乙烯基萘(0.5毫摩尔),糖精(0.5毫摩尔),二苯基二硒醚(0.25毫摩尔),乙腈(2毫升),加毕,充入n2保护,将23瓦白色紧凑型荧光灯放在距离反应管3厘米的地方,室温下反应20小时,反应完成后,将有机相通过旋转蒸发器除去溶剂,残余物用硅胶柱进行纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯=9:1),得到黄色固体194毫克,产率为79%,
所得产物核磁图谱数据为:1hnmr(400mhz,cdcl3):δ/ppm=7.92(s,1h),7.86(dd,j=6.8,1.3hz,1h),7.76–7.58(m,7h),7.47(dd,j=6.3,3.2hz,2h),7.37(dd,j=6.2,3.2hz,2h),7.15(dd,j=5.0,1.7hz,3h),5.48(t,j=8.1hz,1h),4.07(dd,j=12.9,8.1hz,1h),3.85(dd,j=12.9,8.1hz,1h).13cnmr(100mhz,cdcl3):δ/ppm=157.7,136.3,133.7,133.2,132.9,132.7,132.2,131.9,128.1,128.0,127.4,127.3,127.0,126.6,126.5,126.0,125.5,125.3,124.9,124.1,119.7,56.9,28.0。
实施例4
室温下,向装有磁力搅拌子的25毫升schlenk(施兰克)管中装入过2-乙烯基噻吩(0.5毫摩尔),糖精(0.5毫摩尔),二苯基二硒醚(0.25毫摩尔),乙腈(2毫升),加毕,充入n2保护,将23瓦白色紧凑型荧光灯放在距离反应管3厘米的地方,室温下反应20小时,反应完成后,将有机相通过旋转蒸发器除去溶剂,残余物用硅胶柱进行纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯=12:1),得到白色固体186毫克,产率为83%,
所得产物核磁图谱数据为:1hnmr(400mhz,cdcl3):δ/ppm=7.87(d,j=7.2hz,1h),7.78–7.63(m,3h),7.48(dd,j=6.5,3.0hz,2h),7.21–7.13(m,5h),6.87(dd,j=5.1,3.6hz,1h),5.53(t,j=8.0hz,1h),3.99(dd,j=13.0,8.0hz,1h),3.72(dd,j=13.0,8.0hz,1h).13cnmr(100mhz,cdcl3):δ/ppm=157.4,138.1,136.4,133.8,133.2,132.8,128.2,127.7,127.0,126.7,125.9,125.6,125.3,124.2,119.8,51.7,29.4。
实施例5
室温下,向装有磁力搅拌子的25毫升schlenk(施兰克)管中装入过环辛烯(0.5毫摩尔),糖精(0.5毫摩尔),二苯基二硒醚(0.25毫摩尔),乙腈(2毫升),加毕,充入n2保护,将23瓦白色紧凑型荧光灯放在距离反应管3厘米的地方,室温下反应20小时,反应完成后,将有机相通过旋转蒸发器除去溶剂,残余物用硅胶柱进行纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯=10:1),得到白色油状物146毫克,产率为65%,
所得产物核磁图谱数据为:1hnmr(400mhz,cdcl3):δ/ppm=7.90(dd,j=16.5,7.4hz,2h),7.80(dtd,j=22.2,7.4,1.2hz,2h),7.51(dd,j=7.1,2.3hz,2h),7.11(dd,j=5.3,1.7hz,3h),4.60–4.50(m,1h),4.45(d,j=8.1hz,1h),2.41(ddd,j=18.4,10.7,3.4hz,1h),2.34–2.22(m,1h),2.20–2.08(m,1h),1.98–1.61(m,7h),1.54(d,j=11.4hz,2h).13cnmr(100mhz,cdcl3):δ/ppm=157.9,136.2,134.0,133.5,133.0,128.3,127.7,126.4,124.0,119.7,58.2,32.0,28.5,26.4,24.9,24.3,24.2,21.6。
实施例6
室温下,向装有磁力搅拌子的25毫升schlenk(施兰克)管中装入过苯乙烯(0.5毫摩尔),糖精(0.5毫摩尔),二甲基二硒醚(0.25毫摩尔),乙腈(2毫升),加毕,充入n2保护,将23瓦白色紧凑型荧光灯放在距离反应管3厘米的地方,室温下反应20小时,反应完成后,将有机相通过旋转蒸发器除去溶剂,残余物用硅胶柱进行纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯=11:1),得到黄色固体162毫克,产率为85%,
所得产物核磁图谱数据为:1hnmr(400mhz,cdcl3):δ/ppm=8.01(dd,j=6.7,1.4hz,1h),7.90–7.77(m,3h),7.63(d,j=6.9hz,2h),7.42–7.31(m,3h),5.39(t,j=8.2hz,1h),3.68(dd,j=12.9,8.2hz,1h),3.53(dd,j=12.9,8.2hz,1h),2.04(s,3h).13cnmr(100mhz,cdcl3):δ/ppm=157.8,136.4,135.8,133.7,133.3,127.8,127.6,127.5,126.1,124.2,119.8,56.5,25.2,4.1。
实施例7
室温下,向装有磁力搅拌子的25毫升schlenk(施兰克)管中装入过苯乙烯(0.5毫摩尔),糖精(0.5毫摩尔),二苄基二硒醚(0.25毫摩尔),乙腈(2毫升),加毕,充入n2保护,将23瓦白色紧凑型荧光灯放在距离反应管3厘米的地方,室温下反应20小时,反应完成后,将有机相通过旋转蒸发器除去溶剂,残余物用硅胶柱进行纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯=9:1),得到黄色固体192毫克,产率为84%,
所得产物核磁图谱数据为:1hnmr(400mhz,cdcl3):δ/ppm=8.01(dd,j=6.7,1.4hz,1h),7.90–7.76(m,3h),7.53(dd,j=8.0,1.4hz,2h),7.39–7.27(m,8h),5.25(t,j=8.2hz,1h),3.83(d,j=4.6hz,2h),3.64(dd,j=13.1,8.3hz,1h),3.41(dd,j=13.1,8.3hz,1h).13cnmr(100mhz,cdcl3):δ/ppm=157.8,137.6,136.3,135.9,133.7,133.3,128.0,127.7,127.6,127.5,127.4,126.2,125.9,124.2,119.8,56.6,26.7,23.6。
实施例8
室温下,向装有磁力搅拌子的25毫升schlenk(施兰克)管中装入过苯乙烯(0.5毫摩尔),双苯磺酰亚胺(0.5毫摩尔),二苯基二硒醚(0.25毫摩尔),乙腈(2毫升),加毕,充入n2保护,将23瓦白色紧凑型荧光灯放在距离反应管3厘米的地方,室温下反应20小时,反应完成后,将有机相通过旋转蒸发器除去溶剂,残余物用硅胶柱进行纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯=10:1),得到白色固体197毫克,产率为71%,
所得产物核磁图谱数据为:1hnmr(400mhz,cdcl3):δ/ppm=7.69–7.60(m,2h),7.46–7.37(m,2h),7.37–7.30(m,4h),7.31–7.24(m,4h),7.22–7.16(m,8h),4.90(dd,j=11.5,3.4hz,1h),4.80(dd,j=15.2,11.5hz,1h),3.48(dd,j=15.2,3.4hz,1h).13cnmr(100mhz,cdcl3):δ/ppm=137.3,137.0,1345.0,132.5,128.4128.2,128.1127.9,127.6,127.5,127.4,127.0,51.3,45.6。
实施例9
室温下,向装有磁力搅拌子的25毫升schlenk(施兰克)管中装入过苯乙烯(0.5毫摩尔),苯并三氮唑(0.5毫摩尔),二苯基二硒醚(0.25毫摩尔),乙腈(2毫升),加毕,充入n2保护,将23瓦白色紧凑型荧光灯放在距离反应管3厘米的地方,室温下反应20小时,反应完成后,将有机相通过旋转蒸发器除去溶剂,残余物用硅胶柱进行纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯=7:1),得到白色油状物144毫克,产率为76%,所得产物核磁图谱数据为:1hnmr(400mhz,cdcl3):δ/ppm=7.97(dt,j=8.0,1.1hz,2h),7.46–7.32(m,2h),7.33–7.13(m,10h),5.77(dd,j=9.4,5.7hz,1h),4.18(dd,j=13.1,9.4hz,1h),3.75(dd,j=13.1,5.7hz,1h).13cnmr(100mhz,cdcl3):δ/ppm=145.0,137.3,132.6,132.0,128.2,128.0,127.8,127.7,126.7,126.2,125.8,122.9,119.0,108.5,62.6,31.5。
本发明制备方法以烯烃,含氮化合物,二硒醚为原料,以乙腈,二氯甲烷、二甲基甲酰胺,乙酸乙酯中的一种为溶剂,反应温度为室温,白色荧光的光源照射下,高效合成出β-胺基硒醚化合物,该方法与传统的合成方法相比,具有反应条件温和,产率高,官能团兼容性好及原子经济性等优势。
尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
- 还没有人留言评论。精彩留言会获得点赞!