一种多级连续串联反应萃取制备维生素A醋酸酯的装置及方法与流程
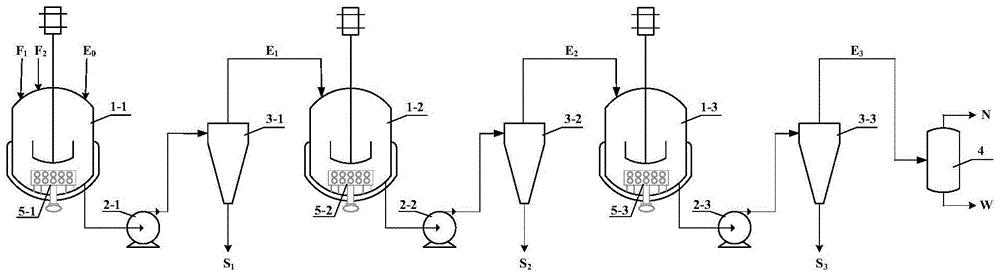
本发明涉及一种多级连续串联反应萃取制备维生素a醋酸酯的装置及方法,属于维生素a及其衍生物生产
技术领域:
。
背景技术:
:维生素a的化学名为视黄醇,是最早被发现的一种脂溶性维生素,是人体必需的营养素之一。但是维生素a非常不稳定,遇酸、空气、氧化性物质、高温或紫外线等容易被破坏,且对皮肤有刺激性。维生素a酯类衍生物较维生素a更加稳定,且不具有刺激性,因此商品维生素a都以酯的形式提供,最常见的商品形式是维生素a醋酸酯。维生素a醋酸酯作为维生素a最主要的系列产品之一,由于其更好的稳定性及更优的药理作用,其需求量日益增大,已广泛应用于化妆品、药物、饲料等。近年来人们对于维生素a醋酸酯的合成研究主要集中于间歇工艺,而维生素a醋酸酯的连续合成工艺鲜有研究。wo2005058811a、de10164041a以及中国专利cn101318975a、cn101219983a和cn102190565a均报道了使用c14醛和c1进行wittig反应,经过步骤繁琐的后处理,得到c15膦酸酯,报道收率为80~92%,但实际低于80%,并且c15膦酸酯必须经过彻底干燥或处理后方能用于下一步wittig反应,如cn1097414a。专利cn103044302a报道了一锅法制备维生素a醋酸酯的方法,其利用c14醛和中间体c1(亚甲基二膦酸四乙酯或亚甲基二膦酸四甲酯)在碱性条件下反应生成c15膦酸酯,该c15膦酸酯不经过分离,直接和c5醛一锅法反应制备维生素a醋酸酯,该方法已经有了很大改进,但仍然避免不了需要使用甲醇钠、乙醇钠、叔丁醇钾或氢化钠等危险且对空气和湿气控制要求高的试剂。专利cn109731612a报道了一种以水为反应介质,在无机碱和功能化离子液体的存在下,c15膦盐与c5醛反应生成全反式维生素a醋酸酯,生产安全更加易于控制,但仍仅适用于间歇生产。以c15膦盐与c5醛为原料生产维生素a醋酸酯,是近年来维生素a醋酸酯合成领域研究的热点,但仍未能实现连续化的生产。反应萃取作为一种过程强化技术,通过将反应和萃取过程耦合,与传统单元操作和其他化工过程强化技术相比,反应萃取技术具有工艺简单、条件温和、经济实用、可设计性强等诸多优点。涉及反应萃取技术在液液反应方面的应用,国内外众多学者进行了大量的研究,而涉及到体系中含有固体的反应,反应萃取技术的运用则相对较少。反应体系中含有固体的反应,在长期运行过程中体系中的固体会沉积在设备底部不易清理,且极易发生堵塞管路的风险,并且由于固体不能及时从反应体系中移出,在搅拌条件下产品、原料、中间体和副产固体间会相互包裹,降低反应速率,影响转化率和收率,增加副反应的发生。在反应萃取工艺中这些问题难以通过搅拌等常规方式解决,不易实现连续化,因此液固相的连续化反应萃取工艺鲜有报道。以c15膦盐和c5醛为反应原料,水为反应溶剂的维生素a醋酸酯制备工艺,在反应过程中会有三苯基氧化膦等副产固体颗粒生成,这些不断生成的固体颗粒,一方面使其实现反应萃取连续化生产制造了困难,一方面也使得副反应增多影响维生素a醋酸酯的合成收率。目前报道的化学合成法制备维生素a醋酸酯的生产工艺均为间歇工艺,存在劳动强度大,生产效率低,对环境影响大等缺点,因此寻求一种合适的富有竞争力的绿色制造工艺成为亟待解决的问题。技术实现要素:针对现有技术中存在的上述问题,本发明对化学合成法合成维生素a醋酸酯的方法进行了改进。本发明公开了一种多级连续串联反应萃取制备维生素a醋酸酯的方法和装置。该方法反应条件温和,实现了连续化生产,生产安全易于控制,环境友好,生成的副产物能够及时移出反应体系,降低了副反应的发生,反应收率高。为了解决上述技术问题,本发明采用的技术方案如下:一种多级连续串联反应萃取制备维生素a醋酸酯的方法,该方法是将原料f1、碱液f2及萃取剂e0持续通入多级连续串联反应萃取装置中进行wittig反应,合成得到维生素a醋酸酯。本发明方法中,所述原料f1为c15膦盐、水和c5醛的混合液。优选地,所述原料f1中,水的用量为c15膦盐质量的1.5~6倍,优选2~5倍;c5醛与c15膦盐的摩尔比为1/1~1.6/1,优选1.03/1~1.2/1。进一步地,所述c15膦盐结构通式为式中x为卤素,优选cl或br,更优选cl,即氯化c15膦盐,其结构式为进一步地,所述c5醛为3-甲酰基丁-2-烯基乙酸酯,其结构式为本发明方法中,所述碱液f2选自氢氧化钠、氢氧化钾、氢氧化锂、碳酸钾、碳酸钠、碳酸铵的水溶液以及氨水中的一种或多种,优选为碳酸钠和/或碳酸钾的水溶液;优选地,所述碱液f2的碱浓度为10~45wt%,优选15~35wt%。进一步地,所述碱液f2中还添加了相转移助剂,所述相转移助剂为季铵盐类相转移助剂,选自三乙基苄基氯化铵、三乙基苄基溴化铵、三辛基甲基溴化铵、三辛基甲基氯化铵、十二烷基三甲基氯化铵、十二烷基三甲基溴化铵、十六烷基三甲基氯化铵、十六烷基三甲基溴化铵、四丁基硫酸氢铵中的一种或多种,优选为十二烷基三甲基氯化铵、十二烷基三甲基溴化铵、十六烷基三甲基氯化铵、十六烷基三甲基溴化铵中的一种或多种;优选地,所述相转移助剂与碱(以化合物计)的摩尔比为1/8~1/50,优选1/15~1/36。本发明在反应体系中加入季铵盐类相转移助剂,一方面可以促进水相碱液中的阴离子与c15膦盐充分接触,有利于中间体叶立德的生成,同时能够继续促进叶立德与c5醛反应生成维生素a醋酸酯,如此可起到缩短反应时间,提高转化率的作用;另一方面,还可以避免反应体系中由于产品、原料、中间体和副产固体间的相互包裹问题带来不利影响,抑制副反应的发生,起到提高选择性的作用。本发明方法中,所述萃取剂e0选自石油醚、正己烷、正庚烷、正戊烷、环己烷、甲基环己烷中的一种或多种,优选石油醚、正己烷、正庚烷中的一种或多种。本发明方法中,所述原料f1与碱液f2的进料质量流量比为(2~20):1;所述原料f1与萃取剂e0的进料质量流量比为(2~4):1。本发明方法中,所述wittig反应机理如下:本发明方法中,所述多级连续串联反应萃取装置,包括多级串联连接的反应萃取重复结构单元和油水分离器,每级反应萃取重复结构单元包含一套依次连接的反应萃取搅拌釜、泵和旋液分离器;所述反应萃取搅拌釜底部设有高速剪切混合器。每一级反应萃取搅拌釜底部增加一个高速剪切混合器,不仅可以使反应体系各相接触更为充分,有利于反应的发生,还能促使反应过程中生成的三苯基氧化膦副产固相分散在液相中,减小了堵塞管路的风险,为连续化生产创造了条件。优选地,所述多级连续串联反应萃取装置的萃取级数为2~6级,优选3~5级。优选地,所述反应萃取搅拌釜中高速剪切混合器的转速为3000~12000rpm,优选5000~9000rpm。本发明方法中,对所述反应萃取搅拌釜中的搅拌不作特别限定,采用常规搅拌即可。实验发现反应萃取法连续生产维生素a醋酸酯的过程中仅通过调整搅拌釜的搅拌速率对解决固体沉积、提高反应速率等并无特别效果,而在反应萃取搅拌釜底部设置高速剪切混合器,其转速对反应效果及产品质量具有显著影响,须控制在特定范围内。若转速控制不当,如过高转速下反应体系易呈乳化状态,会使得三苯基氧膦(tppo)等固相副产过多地停留在液相中,物料相互包裹阻碍反应顺利进行,并为后续分离增加难度;如转速过低,固相易团聚,体系进入管道则易增加堵塞风险,起不到分散固体的作用,无法实现连续化操作。本发明方法中,每级反应萃取搅拌釜中物料的停留时间为2~8min,优选3~6min。本发明方法中,每级反应萃取搅拌釜中的反应温度为35~70℃,优选40~55℃。本发明一些方法中,采用的具体步骤为:将原料f1、碱液f2和萃取剂e0由顶部通入反应萃取搅拌釜中搅拌混合,同时开启高速剪切混合器,进行wittig反应,同时由底部排出反应液,泵送入旋液分离器分离出固体,然后进入下一级萃取重复结构单元重复上述操作,最后进入油水分离器分离得维生素a醋酸酯产品。本发明方法中,高速剪切混合器和季铵盐类相转移助剂的协同作用,使得物料间接触充分,缩短了反应时间,与多级连续串联反应萃取装置中设置的旋液分离器共同作用,能够将反应过程中产生的固相副产物及时从反应体系中移出,避免产品、原料、中间体和副产固体间的相互包裹,有利于减少副反应的发生,同时,旋液分离器为多级连续串联反应萃取装置提供连续的液相,使得多级反应顺利往后进行,实现了固液体系下的连续化生产。本发明还提供一种多级连续串联反应萃取装置,包括多级串联连接的反应萃取重复结构单元和油水分离器,每级反应萃取重复结构单元包含一套依次连接的反应萃取搅拌釜、泵和旋液分离器;所述反应萃取搅拌釜底部设有高速剪切混合器。优选地,所述萃取重复结构单元中,在反应萃取搅拌釜顶部设置进料口,底部设置出料口,出料口通过管线经由泵与旋液分离器入口相连接。优选地,所述多级连续串联反应萃取装置的萃取级数为2~6级。本发明所述多级连续串联反应萃取装置具有普遍适用性,不但可用于液液反应体系萃取工艺,还可用于液固反应体系萃取工艺,尤其适用于本发明有固相副产物生成的维生素a醋酸酯的合成方法。本发明技术方案有益效果在于:(1)本发明的方法实现了维生素a醋酸酯的连续化生产,可以大幅提高生产效率,缩短生产周期,降低生产成本,从本质上也降低了工艺操作劳动强度;(2)反应过程和萃取过程耦合在一起,反应过程产生的固相能够被及时移出反应体系,有利于减少副反应的发生,实现了涉及体系中含有固体的反应萃取连续化工艺生产;(3)经本发明方法得到的维生素a醋酸酯收率高,且方法反应条件温和,环境友好,适合大规模工业化生产,对提高我国维生素a醋酸酯的工业化生产水平有重要意义。附图说明图1为本发明实施例1所采用的三级连续串联反应萃取装置示意图;图中:1-1、1-2、1-3为每一级的反应萃取搅拌釜;2-1、2-2、2-3为每一级的物料输送泵;3-1、3-2、3-3为每一级用于固液分离的旋液分离器;4为用于油水相分离的油水分离器;5-1、5-2、5-3为每一级的反应萃取搅拌釜底部安装的高速剪切混合器;f1为原料(混合液);f2为碱液;e0为萃取剂;e1、e2和e3为每一级经旋液分离器分离后的液相;s1、s2和s3为每一级经旋液分离器分离后的固相;n为经油水分离器分离后的油相(萃取剂相);w为经油水分离器分离后的水相。具体实施方式下面通过实施例对本发明作进一步的阐述,但是这些实施例无论如何都不对本发明的范围构成限制。实施例最后得到的产品通过高效液相色谱分析纯度,用标准样品建立外标曲线,所测得的纯度都是液相外标法的纯度。分析条件:高效液相色谱仪:shimadzulc-20a型号,cto-10asvp柱温箱,spd-m20a检测器,色谱柱odsc18柱(250mm×4.6mm×5μm)。柱温箱温度40℃;流动相为甲醇水溶液(甲醇和水体积比等于3:2);流速为0.4ml/min;uv检测波长为328nm。本发明实施例主要原料来源如表1所示:表1实施例及对比例涉及的试剂来源试剂来源纯度规格氯化c15膦盐自制99.5%溴化c15膦盐自制99.5%吡啶山东昆达生物科技有限公司99.9%乙烯基-β紫罗兰醇浙江新和成股份有限公司94%甲醇山东联盟化工股份有限公司99.9%三苯基膦聊城德丰化工有限公司99.9%盐酸烟台远东精细化工有限公司36~38%氢溴酸烟台远东精细化工有限公司33~35%c5醛(3-甲酰基丁-2-烯基乙酸酯)临沂艾泽拉斯生物科技有限公司99%碳酸钠烟台远东精细化工有限公司99.5%碳酸钾烟台远东精细化工有限公司99%氢氧化钾烟台远东精细化工有限公司99%氢氧化钠烟台远东精细化工有限公司99%氢氧化锂泰安盈锂化工材料有限公司99%碳酸铵烟台远东精细化工有限公司99%氨水烟台远东精细化工有限公司25~28%石油醚烟台远东精细化工有限公司60~90℃沸程正己烷烟台远东精细化工有限公司99%正戊烷烟台远东精细化工有限公司99%正庚烷烟台远东精细化工有限公司99%环己烷烟台远东精细化工有限公司99%甲基环己烷济南谷瑞特化工有限公司99%十六烷基三甲基氯化铵邹平市名兴化工有限公司99%十二烷基三甲基氯化铵邹平市名兴化工有限公司99%十六烷基三甲基溴化铵邹平市名兴化工有限公司99%三乙基苄基氯化铵临沂艾泽拉斯生物科技有限公司99%十二烷基三甲基溴化铵邹平市名兴化工有限公司99%三辛基甲基氯化铵邹平市名兴化工有限公司99%四丁基硫酸氢铵邹平市名兴化工有限公司99%18-冠醚-6临沂艾泽拉斯生物科技有限公司99%其中,c15膦盐制备方法,可参照期刊文献“wittig法合成维生素a新工艺研究[d].吕国锋.浙江工业大学2007”进行。氯化c15膦盐制备过程为:在搅拌釜中加入10.5kg乙烯基-β紫罗兰醇(纯度为94wt%)、2.5l吡啶、25l甲醇,开启搅拌配置混合溶液;往配有冷凝回流装置的搪瓷釜中加入100l甲醇,1.5kg三苯基膦和10l浓度38.0wt%的盐酸水溶液;开启搅拌,对搪瓷釜氮气置换后开始升温,当体系温度达到50℃时,通过泵将搅拌釜中的混合溶液打入搪瓷釜中进行反应,控制加料时间约2h,加料完毕后保温5h;保温结束减压回收溶剂甲醇,回收完后加入乙酸乙酯,搅拌均匀后回收乙酸乙酯。回收完毕后,再次加入乙酸乙酯对产品进行冷却结晶,再抽滤、干燥,即得到氯化c15膦盐(摩尔质量为501.08),其液相色谱纯度大于99.5wt%。溴化c15膦盐制备过程为:参照氯化c15膦盐制备过程,不同之处仅在于将其中的原料“10l浓度38.0wt%的盐酸水溶液”替换为“20.6l浓度35.0wt%的氢溴酸水溶液”,制得溴化c15膦盐(摩尔质量为545.53),其液相色谱纯度大于99.5wt%。本发明实施例采用的三级连续串联反应萃取装置(如图1所示):包括三级串联连接的反应萃取重复结构单元和油水分离器4,每级反应萃取重复结构单元包含一套依次连接的反应萃取搅拌釜1(1~3级分别为1-1、1-2、1-3)、泵2(1~3级分别为2-1、2-2、2-3)和旋液分离器3(1~3级分别为3-1、3-2、3-3);反应萃取搅拌釜1的底部设有高速剪切混合器5(1~3级分别为5-1、5-2、5-3)。每一级萃取重复结构单元中,在反应萃取搅拌釜1顶部设置进料口,底部设置出料口,出料口通过管线经由泵2与旋液分离器3入口相连接。图1中f1为原料(c15膦盐、c5醛和水的混合液);f2为碱液;e0为萃取剂;e1、e2和e3为每一级经旋液分离器3分离后的液相;s1、s2和s3为每一级经旋液分离器3分离后的固相;n为经油水分离器5分离后的油相(萃取剂相);w为经油水分离器分离后的水相。实施例1配制原料f1,其中氯化c15膦盐质量含量为30.37%,c5醛质量含量为8.88%,水质量含量为60.75%(c5醛与氯化c15膦盐的摩尔比为1.03)。原料f1预热至40℃。配制碱液f2,其中碳酸钠质量浓度为15%,相转移助剂十六烷基三甲基氯化铵质量浓度为3%(碳酸钠与十六烷基三甲基氯化铵的摩尔比为15),其余为纯水。碱液f2预热至40℃。选用石油醚作为萃取剂e0。萃取剂e0预热至40℃。反应装置如图1所示,将原料f1、碱液f2和萃取剂e0分别以14.8kg/min、7.0kg/min和7.4kg/min的质量流量同时通入到第一级反应萃取搅拌釜1-1中维持40℃进行反应萃取过程,当釜内持液量为175kg时(控制物料停留时间为6min),开启第一级物料输送泵2-1(控制流量等于原料f1、碱液f2和萃取剂e0的流量之和)和旋液分离器3-1,分出的液相进入下一级反应萃取搅拌釜中继续反应萃取过程,直至得到第三级经旋液分离器分离后的液相e3。第二和第三级反应萃取搅拌釜中进行的反应萃取过程的反应温度和物料停留时间同第一级。每级高速剪切混合器的转速为5000rpm。液相e3进入油水分离器分离后得到油相n(石油醚相)。装置运行稳定后,对得到的油相n进行高效液相色谱分析,维生素a醋酸酯收率为94.44%;装置运行平稳后,5min内从装置中共收集到12.2kg副产固体(其中tppo含量96.62wt%;tppo理论生成量11.8kg,以tppo计算可得固体分离率达99.9%)。实施例2配制原料f1,其中氯化c15膦盐质量含量为15.77%,c5醛质量含量为5.37%,水质量含量为78.86%(c5醛与氯化c15膦盐的摩尔比为1.2)。原料f1预热至55℃。配制碱液f2,其中氢氧化钠质量浓度为35%,相转移助剂十二烷基三甲基氯化铵质量浓度为6.4%,其余为纯水(氢氧化钠与十二烷基三甲基氯化铵的摩尔比为36)。碱液f2预热至55℃。选用正庚烷作为萃取剂e0。萃取剂e0预热至55℃。如附图所示,将原料f1、碱液f2和萃取剂e0分别以38.0kg/min、1.9kg/min和9.4kg/min的质量流量同时通入到第一级反应萃取搅拌釜1-1中维持55℃进行反应萃取过程,当釜内持液量为148kg时(控制物料停留时间为3min),开启第一级物料输送泵2-1(控制流量等于原料f1、碱液f2和萃取剂e0的流量之和)和旋液分离器3-1,分出的液相进入下一级反应萃取搅拌釜中继续反应萃取过程,直至得到第三级经旋液分离器分离后的液相e3。第二和第三级反应萃取搅拌釜中进行的反应萃取过程的反应温度和物料停留时间同第一级。每级高速剪切混合器的转速为9000rpm。液相e3进入油水分离器分离后得到油相n(正庚烷相)。装置运行稳定后,对得到的油相n进行高效液相色谱分析,维生素a醋酸酯收率为94.31%;装置运行平稳后,5min内从装置中共收集到16.2kg副产固体(其中tppo含量96.81wt%;tppo理论生成量15.7kg,以tppo计算可得固体分离率达99.9%以上)。实施例3配制原料f1,其中氯化c15膦盐质量含量为23.19%,c5醛质量含量为7.24%(c5醛与氯化c15膦盐的摩尔比为1.1),水质量含量为69.57%。原料f1预热至50℃。配制碱液f2,其中碳酸钾质量浓度为30%,相转移助剂十六烷基三甲基溴化铵质量浓度为3.2%,其余为纯水(碳酸钾与十六烷基三甲基溴化铵的摩尔比为25)。碱液f2预热至50℃。选用正己烷作为萃取剂e0。萃取剂e0预热至50℃。如附图所示,将原料f1、碱液f2和萃取剂e0分别以21.6kg/min、5.5kg/min和8.7kg/min的质量流量同时通入到第一级反应萃取搅拌釜1-1中维持50℃进行反应萃取过程,当釜内持液量为179kg时(控制物料停留时间为5min),开启第一级物料输送泵2-1(控制流量等于原料f1、碱液f2和萃取剂e0的流量之和)和旋液分离器3-1,分出的液相进入下一级反应萃取搅拌釜中继续反应萃取过程,直至得到第三级经旋液分离器分离后的液相e3。第二和第三级反应萃取搅拌釜中进行的反应萃取过程的反应温度和物料停留时间同第一级。每级高速剪切混合器的转速为7000rpm。液相e3进入油水分离器分离后得到油相n(正己烷相)。装置运行稳定后,对得到的油相n进行高效液相色谱分析,维生素a醋酸酯收率为95.12%;装置运行平稳后,5min内从装置中共收集到13.8kg副产固体(其中tppo含量95.55wt%;tppo理论生成量13.2kg,以tppo计算可得固体分离率达99.9%)。实施例4配制原料f1,其中氯化c15膦盐质量含量为35.92%,c5醛质量含量为10.19%,水质量含量为53.89%(c5醛与氯化c15膦盐的摩尔比为1)。原料f1预热至35℃。配制碱液f2,其中氢氧化锂质量浓度为10%,相转移助剂三乙基苄基氯化铵质量浓度为11.9%,其余为纯水(氢氧化锂与三乙基苄基氯化铵的摩尔比为8)。碱液f2预热至35℃。选用正戊烷作为萃取剂e0。萃取剂e0预热至35℃。如附图所示,将原料f1、碱液f2和萃取剂e0分别以16.7kg/min、3.0kg/min和4.9kg/min的质量流量同时通入到第一级反应萃取搅拌釜1-1中维持35℃进行反应萃取过程,当釜内持液量为197kg时(控制物料停留时间为8min),开启第一级物料输送泵2-1(控制流量等于原料f1、碱液f2和萃取剂e0的流量之和)和旋液分离器3-1,分出的液相进入下一级反应萃取搅拌釜中继续反应萃取过程,直至得到第三级经旋液分离器分离后的液相e3。第二和第三级反应萃取搅拌釜中进行的反应萃取过程的反应温度和物料停留时间同第一级。每级高速剪切混合器的转速为3000rpm。液相e3进入油水分离器分离后得到油相n(正戊烷相)。装置运行稳定后,对得到的油相n进行高效液相色谱分析,维生素a醋酸酯收率为93.84%;装置运行平稳后,5min内从装置中共收集到16.2kg副产固体(其中tppo含量95.91wt%;tppo理论生成量15.6kg,以tppo计算可得固体分离率达99.6%)。实施例5配制原料f1,其中氯化c15膦盐质量含量为13.42%,c5醛质量含量为6.09%,水质量含量为80.49%(c5醛与氯化c15膦盐的摩尔比为1.6)。原料f1预热至70℃。配制碱液f2,其中碳酸铵质量浓度为45%,相转移助剂十二烷基三甲基溴化铵质量浓度为2.9%,其余为纯水(碳酸铵与十二烷基三甲基溴化铵的摩尔比为50)。碱液f2预热至70℃。选用甲基环己烷作为萃取剂e0。萃取剂e0预热至70℃。如附图所示,将原料f1、碱液f2和萃取剂e0分别以52.2kg/min、6.0kg/min和21.8kg/min的质量流量同时通入到第一级反应萃取搅拌釜1-1中维持70℃进行反应萃取过程,当釜内持液量为160kg时(控制物料停留时间为2min),开启第一级物料输送泵2-1(控制流量等于原料f1、碱液f2和萃取剂e0的流量之和)和旋液分离器3-1,分出的液相进入下一级反应萃取搅拌釜中继续反应萃取过程,直至得到第三级经旋液分离器分离后的液相e3。第二和第三级反应萃取搅拌釜中进行的反应萃取过程的反应温度和物料停留时间同第一级。每级高速剪切混合器的转速为12000rpm。液相e3进入油水分离器分离后得到油相n(甲基环己烷相)。装置运行稳定后,对得到的油相n进行高效液相色谱分析,维生素a醋酸酯收率为93.18%;装置运行平稳后,5min内从装置中共收集到18.8kg副产固体(其中tppo含量95.99wt%;tppo理论生成量18.1kg,以tppo计算可得固体分离率达99.7%)。实施例6配制原料f1,其中氯化c15膦盐质量含量为18.63%,c5醛质量含量为6.87%,水质量含量为74.50%(c5醛与氯化c15膦盐的摩尔比为1.3)。原料f1预热至60℃。配制碱液f2,其中氨水质量浓度为25%,相转移助剂三辛基甲基氯化铵质量浓度为10.7%,其余为纯水(氨水与三辛基甲基氯化铵的摩尔比为27)。碱液f2预热至60℃。选用环己烷作为萃取剂e0。萃取剂e0预热至60℃。如附图所示,将原料f1、碱液f2和萃取剂e0分别以24.2kg/min、1.9kg/min和9.2kg/min的质量流量同时通入到第一级反应萃取搅拌釜1-1中维持60℃进行反应萃取过程,当釜内持液量为141kg时(控制物料停留时间为4min),开启第一级物料输送泵2-1(控制流量等于原料f1、碱液f2和萃取剂e0的流量之和)和旋液分离器3-1,分出的液相进入下一级反应萃取搅拌釜中继续反应萃取过程,直至得到第三级经旋液分离器分离后的液相e3。第二和第三级反应萃取搅拌釜中进行的反应萃取过程的反应温度和物料停留时间同第一级。每级高速剪切混合器的转速为6000rpm。液相e3进入油水分离器分离后得到油相n(环己烷相)。装置运行稳定后,对得到的油相n进行高效液相色谱分析,维生素a醋酸酯收率为93.92%;装置运行平稳后,5min内从装置中共收集到12.3kg副产固体(其中tppo含量95.02wt%;tppo理论生成量11.7kg,以tppo计算可得固体分离率为99.9%以上)。实施例7配制原料f1,其中溴化c15膦盐质量含量为23.33%,c5醛质量含量为6.69%,水质量含量为69.99%(c5醛与溴化c15膦盐的摩尔比为1.1)。原料f1预热至50℃。配制碱液f2,其中氢氧化钾质量浓度为30%,相转移助剂四丁基硫酸氢铵质量浓度为7.3%,其余为纯水(氢氧化钾与四丁基硫酸氢铵的摩尔比为25)。碱液f2预热至50℃。选用正己烷作为萃取剂e0。萃取剂e0预热至50℃。如附图所示,将原料f1、碱液f2和萃取剂e0分别以23.6kg/min、2.3kg/min和8.7kg/min的质量流量同时通入到第一级反应萃取搅拌釜1-1中维持50℃进行反应萃取过程,当釜内持液量为173kg时(控制物料停留时间为5min),开启第一级物料输送泵2-1(控制流量等于原料f1、碱液f2和萃取剂e0的流量之和)和旋液分离器3-1,分出的液相进入下一级反应萃取搅拌釜中继续反应萃取过程,直至得到第三级经旋液分离器分离后的液相e3。第二和第三级反应萃取搅拌釜中进行的反应萃取过程的反应温度和物料停留时间同第一级。每级高速剪切混合器的转速为7000rpm。液相e3进入油水分离器分离后得到油相n(正己烷相)。装置运行稳定后,对得到的油相n进行高效液相色谱分析,维生素a醋酸酯收率为94.24%;装置运行平稳后,5min内从装置中共收集到13.6kg副产固体(其中tppo含量96.86wt%;tppo理论生成量13.2kg,以tppo计算可得固体分离率达99.8%)。实施例8方法参照实施例3,不同之处仅在于:每级高速剪切混合器的转速调整为2500rpm。维生素a醋酸酯收率为85.95%;装置运行平稳后,5min内从装置中共收集到12.4kg副产固体(其中tppo含量93.28wt%;tppo理论生成量11.9kg,以tppo计算可得固体分离率约97.2%)。实施例9方法参照实施例3,不同之处仅在于:每级高速剪切混合器的转速调整为12500rpm。维生素a醋酸酯收率为87.37%;装置运行平稳后,5min内从装置中共收集到12.1kg副产固体(其中tppo含量96.90wt%;tppo理论生成量12.1kg,以tppo计算可得固体分离率达96.9%)。对比例1方法参照实施例3,不同之处仅在于:碱液f2组成中不加相转移助剂十六烷基三甲基溴化铵,其中碳酸钾质量浓度为30%,其余为纯水。维生素a醋酸酯收率为83.24%;装置运行平稳后,5min内从装置中共收集到11.7kg副产固体(其中tppo含量98.25wt%;tppo理论生成量11.6kg,以tppo计算可得固体分离率为99.1%)。对比例2方法参照实施例3,不同之处仅在于:碱液f2中相转移助剂替换为“18-冠醚-6”。维生素a醋酸酯收率为84.34%;装置运行平稳后,5min内从装置中共收集到12.0kg副产固体(其中tppo含量97.40wt%;tppo理论生成量11.7kg,以tppo计算可得固体分离率为99.9%)。对比例3方法参照实施例3,不同之处仅在于:图1所示装置中,反应萃取搅拌釜底部不设置高速剪切混合器,将原料f1、碱液f2和萃取剂e0分别以21.6kg/min、5.5kg/min和8.7kg/min的质量流量同时通入到第一级反应萃取搅拌釜1-1中维持50℃进行反应萃取过程,当釜内持液量为179kg时(控制物料停留时间为5min),开启第一级物料输送泵2-1(控制流量等于原料f1、碱液f2和萃取剂e0的流量之和)和旋液分离器3-1,装置运行3min后泵2-1入口发生堵塞,无法进行连续生产。对比例4方法参照实施例3,不同之处仅在于:图1所示装置为间歇反应萃取,即采用一套反应萃取搅拌釜、泵和旋液分离器。将原料f1、碱液f2和萃取剂e0分别以21.6kg/min、5.5kg/min和8.7kg/min的质量流量同时通入到第一级反应萃取搅拌釜1-1中,当釜内持液量为179kg时,停止进料,釜内维持50℃反应萃取15min后(高速剪切混合器5-1的转速为7000rpm),开启物料输送泵2-1和旋液分离器3-1,液相e1直接进入油水分离器4分离后得到油相n(正己烷相),对其进行高效液相色谱分析,维生素a醋酸酯收率为61.33%;反应完后从装置中共收集到1.8kg副产固体(其中tppo含量87.27wt%;tppo理论生成量1.7kg,以tppo计算可得固体分离率92.4%)。对比例5方法参照实施例3,不同之处仅在于:图1所示装置为一级连续反应萃取,即仅采用一套反应萃取搅拌釜、泵和旋液分离器。将原料f1、碱液f2和萃取剂e0分别以7.3kg/min、1.9kg/min和2.9kg/min的质量流量同时通入到第一级反应萃取搅拌釜1-1中维持50℃进行反应萃取过程(高速剪切混合器5-1的转速为7000rpm),当釜内持液量为182kg时(控制物料停留时间为15min),开启第一级物料输送泵2-1(控制流量等于原料f1、碱液f2和萃取剂e0的流量之和)和旋液分离器3-1,分出的液相e1直接进入油水分离器分离后得到油相n(正己烷相)。装置运行稳定后,对得到的油相n进行高效液相色谱分析,维生素a醋酸酯收率为68.57%;装置运行平稳后,5min内从装置中共收集到3.4kg副产固体(其中tppo含量89.32wt%;tppo理论生成量3.2kg,以tppo计算可得固体分离率达94.9%)。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1