一种羧甲基酵母β-葡聚糖-表没食子儿茶素没食子酸酯衍生物的制作方法
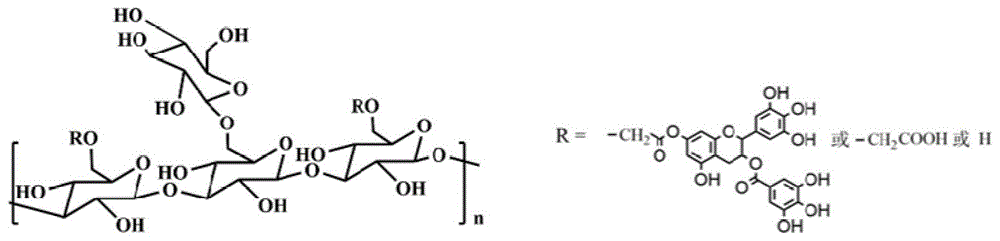
本发明涉及一种羧甲基酵母β-葡聚糖-表没食子儿茶素没食子酸酯衍生物,属于生物质材料技术领域。
背景技术:
类黄酮又称生物类黄酮,为人类饮食中含量最丰富的一类多酚化合物,广泛存于水果、蔬菜、谷物、根茎、树皮、花卉、茶叶和红葡萄酒中,这些物质由于存在性质不稳定限制应用范围,比如表没食子儿茶素-3-没食子酸酯(egcg),是绿茶所含儿茶素中含量最高的成分,是表没食子儿茶素的酯类衍生物。因其独特的类黄酮立体化学结构和多个酚羟基的加持,使得自身具有抗氧化、抗菌、抗炎、抗癌等多种生物活性,可以用于食品工业、医药工业、日化产品中。由于存在化学性质不稳定,见光变色等问题限制应用范围。
目前已有通过将多糖结合类黄酮对类黄酮进行改性,主要接枝多糖的方法有化学偶联法、酶催化接枝法、自由基介导接枝法、酸催化缩合法。酶催化接枝法主要基于黄酮类化合物上羟基的氧化,这样可能会导致嫁接产物中的黄酮类化合物部分功能丧失;自由基介导接枝法的反应效率低,并且使用过程中的过氧化氢还有可能会导致多糖的降解,降低共轭物的分子量;酸催化缩合过程中使用到的高碘酸钠是一种强氧化剂,且有毒,它不仅可以氧化多糖和类黄酮,还可能危害人的健康。近年来,maríaj等人报道的egcg接枝壳聚糖(moreno-vásquezmj,valenzuela-buitimeael,plascencia-jatomeam,etal.functionalizationofchitosanbyafreeradicalreaction:characterization,antioxidantandantibacterialpotential[j].carbohydratepolymers,2017,155:117-127.);fanl等人研究的egcg接枝透明质酸(leef,limj,reithofermr,etal.synthesisandbioactivityofaconjugatecomposedofgreenteacatechinsandhyaluronicacid[j].polymerchemistry,2015,6(24):4462-4472.),二者都是将egcg接枝到氨基上,来提高自由基清除抗氧化效果。然而,目前类黄酮结构物质在光稳定性方面的还存在很多应用限制,见光不稳定易产生褐变。
技术实现要素:
技术问题:
本发明的目的是:①获得生物活性更优异的表没食子儿茶素的酸酯类物质,例如强抗氧化活性、抗菌活性、抗癌活性、抑酶作用等,从而可以应用于更多领域;提高产率;②提高egcg的光稳定性,比如解决光老化问题,延长它的使用寿命。
技术方案:
提供一种制备羧甲基酵母β-葡聚糖-表没食子儿茶素没食子酸酯衍生物的方法,所述方法包括如下步骤:
(1)将羧甲基酵母β-葡聚糖(cmg)分散于溶剂中,然后加入羰基活化剂进行活化处理;
(2)然后在0-10℃下加入表没食子儿茶素没食子酸酯,混匀后,升温至30℃-35℃进行缩合反应,反应结束后,即得羧甲基酵母β-葡聚糖-表没食子儿茶素没食子酸酯衍生物,记为cmg-egcg。
在本发明的一种实施方式中,步骤(1)所述溶剂为dmso与水的混合溶液。其中,dmso与水的体积比为(40-70):100(v/v)。优选(40-55):100。
在本发明的一种实施方式中,步骤(1)中cmg相对溶剂的浓度为14-21mg/ml。
在本发明的一种实施方式中,羧甲基酵母β-葡聚糖的重复单元分子与表没食子儿茶素没食子酸酯的摩尔比为1:(2.0-4.5)。
在本发明的一种实施方式中,步骤(1)所述羰基活化剂为1-(3-二甲基氨基丙基)碳二亚胺盐酸盐(edc)和4-二甲氨基吡啶(dmap)。
在本发明的一种实施方式中,cmg的重复单元分子、edc、dmap的摩尔比为1:
(4.5-5.0):(0.7-1.5)。
在本发明的一种实施方式中,所述羧甲基酵母β-葡聚糖的重复单元分子的结构如下所示:
在本发明的一种实施方式中,步骤(2)中表没食子儿茶素没食子酸酯的加入,是将egcg溶于dmso中,再逐滴加入。
在本发明的一种实施方式中,将egcg溶于dmso中,配置成0.15-0.20g/ml的溶液进行滴加。
在本发明的一种实施方式中,缩合反应结束后,利用乙醇、甲醇洗涤多次,然后固液分离,收集沉淀,并干燥,得到淡黄色固体,即为cmg-egcg。
在本发明的一种实施方式中,干燥是在50℃真空干燥箱中干燥24h。
在本发明的一种实施方式中,具体的制备过程如下:
(1)将一定量的cmg溶于二甲亚砜-水溶液,加入1-(3-二甲基氨基丙基)碳二亚胺盐酸盐(edc)和4-二甲氨基吡啶(dmap),混匀搅拌10min;
(2)取egcg溶于dmso溶液中,再逐滴加入反应体系,冷水浴5min;将温度升至30℃-35℃,继续搅拌16h;反应结束,依次用乙醇、甲醇洗涤,该操作重复三次,将粉末置于50℃真空干燥箱中,干燥24h,得到淡黄色固体,即为羧甲基酵母β-葡聚糖-表没食子儿茶素没食子酸酯衍生物,记为样品cmg-egcg。
本发明考虑到活化羧基的时间(会导致取代度低和产率低),在实验过程的第二步中,向反应体系加入1-(3-二甲基氨基丙基)碳二亚胺盐酸盐(edc)和4-二甲氨基吡啶(dmap)后,等待约10min以活化羧基;另外,当dmso与水混合时会产生局部过热问题,局部过热有可能会使部分egcg被氧化,因此,在实验过程的第三步中,向反应体系滴入egcg的dmso溶液后,冷却了约5min。
本发明利用上述制备方法提供一种羧甲基酵母β-葡聚糖-表没食子儿茶素没食子酸酯衍生物。
所述羧甲基酵母β-葡聚糖-表没食子儿茶素没食子酸酯衍生物的结构如下所示:
其中,n为300-330。
在本发明的一种实施方式中,羧甲基酵母β-葡聚糖-表没食子儿茶素没食子酸酯衍生物的接枝率为20-50mg/g。
本发明还提供一种改善表没食子儿茶素没食子酸酯衍生物稳定性或者抗氧化性的方法,所述方法包括:将羧甲基酵母β-葡聚糖(cmg)分散于溶剂中,然后加入羰基活化剂进行活化处理;然后在0-10℃下加入表没食子儿茶素没食子酸酯,混匀后,升温至30℃-35℃进行缩合改性。
本发明还将上述羧甲基酵母β-葡聚糖-表没食子儿茶素没食子酸酯衍生物应用于食品工业、医药工业、日化领域中。
有益效果:
本发明采用的化学偶联法是edc介导的酯化反应,并对其过程进行了优化,考虑到了温度和反应投料顺序的问题,制得的羧甲基酵母β-葡聚糖-表没食子儿茶素没食子酸酯衍生物的接枝效率较高,为20-50mg/g。
本发明羧甲基酵母β-葡聚糖-表没食子儿茶素没食子酸酯衍生物(cmg-egcg)较羧甲基酵母β-葡聚糖(cmg)有更强的抗氧化性,cmg-egcg对abts自由基的抑制率在浓度为1.0mg/ml时,它的抑制率为95%,而cmg只有6%。cmg-egcg较egcg有更好的颜色稳定性,本发明分别在光照、ph两个不同条件下研究了egcg和cmg-egcg水溶液的颜色变化,发现egcg变色较快且明显,而cmg-egcg变色缓慢。cmg-egcg水溶液在光照15天后无明显的颜色变化;在ph值为7时静置20天,溶液颜色也无明显变化。
附图说明
图1为cmg-egcg衍生物的分子结构式。
图2为cmg、egcg和实施例1所得cmg-egcg的红外光谱图。
图3为egcg和实施例1所得cmg-egcg衍生物自然光照后的颜色对比图。
图4为egcg和实施例1所得cmg-egcg溶液受ph值影响的颜色对比图。
图5为cmg、egcg和实施例1所得cmg-egcg的abts自由基清除率图。
图6为经30天光照后的egcg对abts自由基清除率图。
具体实施方式
实施例1:
取0.87g羧甲基酵母葡聚糖(cmg,以重复单元结构计,为1.14mmol)溶于60ml50%(v/v)二甲亚砜(dmso)水溶液;加入1g1-(3-二甲基氨基丙基)碳二亚胺盐酸盐(edc)和0.2g4-二甲氨基吡啶(dmap),等待10min以活化羧基;
取2.17gegcg(4.7mmol,与羧甲基酵母葡聚糖重复单元结构的摩尔比为4:1)(10mldmso)逐滴加入反应体系中,冷水浴5min;将温度升至30℃-35℃,继续搅拌16h,反应结束,依次用250ml乙醇、50ml甲醇洗涤,该操作重复三次,将过滤得到的滤渣置于50℃真空干燥箱中,干燥24h,得到淡黄色固体,即为羧甲基β-葡聚糖-表没食子儿茶素没食子酸酯衍生物,记为样品cmg-egcg。
图1为cmg-egcg衍生物的分子结构式。egcg接枝率的测定,具体操作如下:将1ml样品水溶液稀释到适当浓度,加入1ml稀释3倍之后的folin-ciocalteu试剂,加入2ml28%(w/v)碳酸钠溶液,充分混匀2min,再加入2ml去离子水,测试总体积为6ml,静置30min后,在760nm处测试吸光度。以egcg水溶液(浓度范围是0.005至0.05mg/ml)为标样,得到接枝率的计算公式:y=17.05x+0.01583(r2=0.9922),其中,x代表了接枝的egcg浓度(mg/ml),y代表了混合溶液在760nm处的吸光度,聚合物的接枝率由每克样品中所含egcg毫克数当量表示。测得本实施例中cmg-egcg的egcg接枝率为29mg/g。
图2为cmg、cmg-egcg、egcg的红外光谱图。从cmg的谱图中可看到位于1733cm-1处的羧酸的特征峰,对应于官能团c=o的伸缩振动;与cmg的红外谱图相比较,cmg-egcg的红外谱图中羧酸特征峰减弱,并分别出现了位于1674cm-1、1589cm-1、1141cm-1新的特征峰,分别对应于酯的c=o的伸缩振动,芳香族c=c的伸缩振动、醇c-oh的伸缩振动(结构归属可参见文献leebs,leecc,linhp,etal.afunctionalchitosanmembranewithgraftedepigallocatechin-3-gallateandlovastatinenhancesperiodontaltissueregenerationindogs[j].carbohydratepolymers,2016,151:790-802.),由此可知,cmg和egcg确已偶联成功。
图3为egcg和cmg-egcg衍生物的水溶液受自然光照一天之后的颜色对比图。egcg和cmg-egcg浓度都是0.6mg/ml。a为未受光照的egcg和产品,b、c分别为经1天、15天自然光照后的egcg和产品,其中左边为egcg水溶液,右边为cmg-egcg衍生物的水溶液。由图可知,经1天光照后,左边的egcg已经发生明显的变色,从未受光照前的透明无色变为淡黄色,而右边的cmg-egcg衍生物水溶液没有明显的变色;15天后,egcg水溶液已经完全变成了黄色,而cmg-egcg衍生物才开始出现变色趋势。
图4对比了egcg和cmg-egcg衍生物在ph值为7时静置20天后的溶液颜色。配制0.6mg/ml的egcg水溶液、cmg-egcg水溶液,并分别调节水溶液的ph至7。其中左边为egcg水溶液,右边为cmg-egcg衍生物的水溶液。由图4可知,egcg和cmg-egcg水溶液的初始颜色都未发生明显变化;但在静置20天后,egcg水溶液的颜色由无色变为了黄色,而cmg-egcg水溶液的颜色几乎无变化。
abts自由基的抑制率测得过程:
取10ml7mmol/labts水溶液与10ml4.9mmol/lk2s2o8溶液混合,在室温、黑暗状态下静置15h,制备abts自由基溶液。用10mmol/l磷酸盐缓冲液(pbs,ph7.4)调节abts+溶液在734nm时的吸光度为0.70±0.05。配置0.2-1mg/ml浓度范围内的5个梯度(0.2mg/ml、0.4mg/ml、0.6mg/ml、0.8mg/ml、1.0mg/ml)的样品水溶液,取200μl各样品溶液与3mlabts+溶液混匀,反应60min,在734nm处测吸光度,计算公式:
abts自由基清除率(%)=(abs0-abs1)/abs0×100
其中abs1、abs0分别指代含样品时abts+溶液的吸光度和空白的abts+溶液的吸光度。
图5显示了egcg、cmg-egcg和na-cmg对abts自由基抑制趋势。由图5可知,egcg表现出最好的abts自由基抑制效果,在浓度为0.2mg/ml时,就已达到99.83%的抑制率,但稳定性差。cmg-egcg与na-cmg对abts自由基的抑制率都与浓度相关,且浓度越高,抑制效果就越好,但cmg-egcg在浓度为0.6mg/ml时,对abts自由基的抑制率已经达到95.2%,之后(0.6-1.0mg/ml)趋于平稳,在为1.0mg/ml时,它的抑制率为95%。而na-cmg样品在浓度为1.0mg/ml时,对abts自由基的抑制率也只有6.05%,远远低于cmg-egcg。
此外,参照现有方法(moreno-vásquezmj,etal.functionalizationofchitosanbyafreeradicalreaction:characterization,antioxidantandantibacterialpotential[j].carbohydratepolymers,2017,155:117-127.)制备的egcg接枝壳聚糖产品,记作壳聚糖-egcg。在浓度为1.0mg/ml时,对abts自由基的抑制率约为40%。
说明本实施例中egcg与cmg接枝后,二者结构存在配合作用,使得具备更优异抗氧化活性的同时,还能够具有较好的光稳定性。
图6显示出了经30天光照后的egcg对abts自由基抑制趋势。由图可知,在0.2-1.0mg/ml浓度范围内,egcg对abts自由基的抑制率都约为96%,与新鲜配置的egcg水溶液对abts自由基的抑制率几乎相同。可见,光稳定性不受抗氧化活性的影响,具体机制正在进一步探究。
实施例2:
取0.87g羧甲基酵母葡聚糖(cmg,以重复单元结构计,为1.14mmol)溶于60ml40%(v/v)二甲亚砜(dmso)水溶液;加入1g1-(3-二甲基氨基丙基)碳二亚胺盐酸盐(edc)和0.1g4-二甲氨基吡啶(dmap),等待10min以活化羧基;
取1.47gegcg(3.21mmol,与羧甲基酵母葡聚糖重复单元结构的摩尔比为2.8:1)(10mldmso)逐滴加入反应体系中,冷水浴5min。将温度升至30℃-35℃,继续搅拌16h,反应结束,依次用乙醇、甲醇洗涤,该操作重复三次,将过滤得到的滤渣置于50℃真空干燥箱中,干燥24h,得到淡黄色固体,即为羧甲基β-葡聚糖-表没食子儿茶素没食子酸酯衍生物,该衍生物的接枝率约为23mg/g。
光稳定性:将所得cmg-egcg产品配置成0.6mg/ml水溶液,受自然光照1天无明显变化,光照15天后,水溶液才开始出现变色趋势。将所得cmg-egcg产品配置成0.6mg/ml水溶液,调节ph值为7,静置20天后的溶液颜色基本不发生变化。
抗氧化性:在浓度为0.6mg/ml时,对abts自由基的抑制率为87.7%。
实施例3:
取0.87g羧甲基酵母葡聚糖(cmg,以重复单元结构计,为1.14mmol)溶于60ml55%(v/v)二甲亚砜(dmso)水溶液;加入1g1-(3-二甲基氨基丙基)碳二亚胺盐酸盐(edc)和0.2g4-二甲氨基吡啶(dmap),等待10min以活化羧基;
取2.17gegcg(4.7mmol,与羧甲基酵母葡聚糖重复单元结构的摩尔比为4:1)(10mldmso)逐滴加入反应体系中,冷水浴5min。将温度升至30℃-35℃,继续搅拌24h,反应结束,依次用乙醇、甲醇洗涤,该操作重复三次,将过滤得到的滤渣置于50℃真空干燥箱中,干燥24h,得到土黄色固体,即为羧甲基β-葡聚糖-表没食子儿茶素没食子酸酯衍生物cmg-egcg,该衍生物的接枝率约为25mg/g。
光稳定性:将所得cmg-egcg产品配置成0.6mg/ml水溶液,受自然光照1天无明显变化,光照15天后,水溶液才开始出现变色趋势。将所得cmg-egcg产品配置成0.6mg/ml水溶液,调节ph值为7,静置20天后的溶液颜色基本不发生变化。
抗氧化性:在浓度为0.6mg/ml时,对abts自由基的抑制率为91.4%。
实施例4:
取0.87g羧甲基酵母葡聚糖(cmg,以重复单元结构计,为1.14mmol)溶于60ml50%(v/v)二甲亚砜(dmso)水溶液;加入1g1-(3-二甲基氨基丙基)碳二亚胺盐酸盐(edc)和0.2g4-二甲氨基吡啶(dmap),等待10min以活化羧基;
取2.17gegcg(4.7mmol,与羧甲基酵母葡聚糖重复单元结构的摩尔比为4.12:1)(10mldmso)逐滴加入反应体系中,冷水浴5min。将温度升至30℃-35℃,继续搅拌30h,反应结束,依次用乙醇、甲醇洗涤,该操作重复三次,将过滤得到的滤渣置于50℃真空干燥箱中,干燥24h,得到棕黄色固体,即为羧甲基β-葡聚糖-表没食子儿茶素没食子酸酯衍生物,该衍生物的接枝率约为21mg/g。
光稳定性:将所得cmg-egcg产品配置成0.6mg/ml水溶液,受自然光照1天无明显变化,光照15天后,水溶液才开始出现变色趋势。将所得cmg-egcg产品配置成0.6mg/ml水溶液,调节ph值为7,静置20天后的溶液颜色基本不发生变化。
抗氧化性:在浓度为0.6mg/ml时,对abts自由基的抑制率为81.2%。
实施例5:
取0.87g羧甲基酵母葡聚糖(cmg,以重复单元结构计,为1.14mmol)溶于60ml50%(v/v)二甲亚砜(dmso)水溶液;加入1g1-(3-二甲基氨基丙基)碳二亚胺盐酸盐(edc)和0.2g4-二甲氨基吡啶(dmap),等待10min以活化羧基;
取1.08gegcg(2.36mmol,与羧甲基酵母葡聚糖重复单元结构的摩尔比为2:1)(10mldmso)逐滴加入反应体系中,冷水浴5min。将温度升至30℃-35℃,继续搅拌24h,反应结束,依次用乙醇、甲醇洗涤,该操作重复三次,将过滤得到的滤渣置于50℃真空干燥箱中,干燥24h,得到棕黄色固体,即为羧甲基β-葡聚糖-表没食子儿茶素没食子酸酯衍生物,该衍生物的接枝率约为14mg/g。
所得cmg-egcg在浓度为0.6mg/ml时,对abts自由基的抑制率为43%。
实施例6:
取0.87g羧甲基酵母葡聚糖(cmg,以重复单元结构计,为1.14mmol)溶于60ml50%(v/v)二甲亚砜(dmso)水溶液;加入1g1-(3-二甲基氨基丙基)碳二亚胺盐酸盐(edc)和0.1g4-二甲氨基吡啶(dmap),等待10min以活化羧基;
取1.08gegcg(2.36mmol,与羧甲基酵母葡聚糖重复单元结构的摩尔比为2:1)(10mldmso)逐滴加入反应体系中,冷水浴5min。将温度升至30℃-35℃,继续搅拌24h,反应结束,依次用乙醇、甲醇洗涤,该操作重复三次,将过滤得到的滤渣置于50℃真空干燥箱中,干燥24h,得到棕黄色固体,即为羧甲基β-葡聚糖-表没食子儿茶素没食子酸酯衍生物,该衍生物的接枝率约为14mg/g。
所得cmg-egcg在浓度为0.6mg/ml时,对abts自由基的抑制率为43%。
- 还没有人留言评论。精彩留言会获得点赞!