光交联水凝胶材料的制备、原料、产品及应用的制作方法
光交联水凝胶材料的制备、原料、产品及应用
[0001]
本申请是分案申请,本分案申请的母案申请日为2017/11/15,申请号为2017111324722,发明名称为“光交联水凝胶材料的制备、原料、产品及应用”。
技术领域
[0002]
本发明属于生物材料领域,具体涉及一种光交联水凝胶材料的制备、原料、产品及应用。
背景技术:
[0003]
水凝胶是一类高度含水的具有三维网络交联结构的聚合物材料,由于其具有优异的生物相容性及一定的机械强度,可以高度拟合生物组织的微环境,因此广泛应用于组织工程和再生医学。在临床应用中,原位固化的水凝胶具有优异的组织赋型能力,当前,可原位固化的水凝胶根据成胶机理的不同主要有温敏型、双组份注射型、光敏型等。温敏型主要是通过低温下为液相的凝胶前体,到达体内后在体温作用下发生相转变凝胶化实现原位固化(如legoo,羟丁基壳聚糖等),此类水凝胶通常存在凝胶强度弱,温度响应慢且在体内降解慢等问题。双组份注射型主要是将分别含有活性反应官能团的凝胶前体通过双组份注射器边混合边挤出的方式实现原位固化(如fibrin glue,adherus autospray等),因此对活性官能团的交联速度具有很高的要求,成胶速度太慢往往导致凝胶前体被体内的血液或渗液稀释或冲走,成胶速度太快则不利于临床操作,易堵针头,同时双组份注射器较为昂贵,大大增加了其应用成本,以上缺陷限制了这类材料的广泛应用。
[0004]
光敏型水凝胶相对于温敏和双组份水凝胶,由于具有时空精确可控的优势而更具备临床的实际操作性。就当前光交联制备水凝胶的方式而言,通过自由基引发不饱和生物大分子聚合交联是当前最为通用的方法。虽然光引发自由基聚合交联的方式固化速度快(2s 左右),但自由基不可避免会对细胞或生物组织造成损伤,且自由基与生俱来的氧气阻聚使得用该方法原位构筑薄层水凝胶十分困难,同时,该类水凝胶与组织粘附能力缺失也一直是该技术临床转化的壁垒。目前为止,fda批准了唯一一例光敏水凝胶-focalseal,用于防止肺切除术后的疝气形成,就在最近biomet公司收购了john hopkins大学的光原位水凝胶构建技术,用于软骨组织修复。虽然以上技术都取得了非常好的临床效果,但它们的使用都必需配合额外底涂,以促进凝胶与组织附着,使得光敏水凝胶的临床使用复杂化。
[0005]
针对光引发自由基聚合交联制备水凝胶技术的不足,朱麟勇研究组于2014年提出了非自由基光偶合交联技术(yunlong yang;jieyuan zhang;zhenzhen liu;qiuning lin; xiaolin liu;chunyan bao;yang wang;linyong zhu.adv.mater.2016,28,2724.;linyongzhu et.al.pct.no.wo2016082725 a1,issued jun2,2016),该技术基于邻硝基苄醇受紫外光照产生醛基,进一步交联多胺基高分子衍生物制备水凝胶,完全规避了自由基的产生,能够有效解决自由基的毒性以及氧阻聚,且胶层薄厚可调,同时,邻硝基苄醇受光照产生的醛基也会与组织表面富含的蛋白胺基交联,实现胶层与组织的化学键键连固定,解决传统光敏型水凝胶的组织粘附与整合等难题。但该技术成胶速度较慢,限制了其临床应用。
技术实现要素:
[0006]
本发明第一个目的是提供环状邻硝基苄基类光扳机,如结构式i-2所示
[0007][0008]
式i-2中,x为o、s或n,x=o时,为环状邻硝基苄基类光扳机,x=s时,为环状邻硝基苄硫基类光扳机,x=n时,为环状邻硝基苄胺基类光扳机;
[0009]
式
ⅰ-
2中,连接键r1一端与x连接,另一端与r2,r3,r4,r5中任意的一个基团相连接,构成环状结构;
[0010]
式i-2中,r
’
选自氢、卤原子、羟基、巯基、胺基、硝基、氰基、醛基、酮基、酯基、酰胺基、膦酸基、膦酸酯基、磺酸基、磺酸酯基、砜基、亚砜基、芳基、杂芳基、烷基、亚烷基、改性烷基或改性亚烷基等;
[0011]
式i-2中,r1选自氢、醚键类取代基、酯键类取代基、碳酸酯键类取代基、胺基甲酸酯键类取代基、巯基甲酸酯键类取代基或磷酸酯键类取代基等;
[0012]
式i-2中,r2,r3,r4,r5可自由的选自氢、卤原子、羟基、巯基、胺基、硝基、氰基、醛基、酮基、羧基、酯基、酰胺基、膦酸基、膦酸酯基、磺酸基、磺酸酯基、砜基、亚砜基、芳基、杂芳基、烷基、亚烷基、改性烷基或改性亚烷基等。
[0013]
对于上述环状邻硝基苄基类光扳机,r2,r3,r4,r5可以相互连接,与碳原子一起形成饱和或不饱和的脂环或脂杂环,或形成芳环或芳杂环。
[0014]
进一步地,所述烷基为具有1~30个碳原子的饱和或不饱和脂肪族直链或支链的烷基;
[0015]
所述亚烷基为具有1~30个碳原子的饱和或不饱和脂肪族直链或支链的亚烷基;
[0016]
所述改性烷基为烷基的任意碳原子被选自卤原子、-oh、-sh、-no2、-cn、-cho、
ꢀ-
cooh、酯基、酰胺基、芳基、亚芳基、-co-、-o-、-s-、-so-、-so
2-、伯胺基、仲胺基、叔胺基、季铵盐基、饱和或不饱和的单环或双环亚环烃基、桥联脂杂环中的至少一种基团置换所得的基团,所述改性烷基具有1~30个原子,其碳碳单键可任意地被碳碳双键或碳碳叁键替换;
[0017]
所述改性亚烷基为亚烷基的任意碳原子被选自卤原子、-oh、-sh、-no2、-cn、-cho、
ꢀ-
cooh、酯基、酰胺基、芳基、亚芳基、-co-、-o-、-s-、-so-、-so
2-、伯胺基、仲胺基、叔胺基、季铵盐基、饱和或不饱和的单环或双环亚环烃基、桥联脂杂环中的至少一种基团置换所得的基团,所述改性亚烷基具有1~30个原子,其碳碳单键可任意地被碳碳双键或碳碳叁键替换;
[0018]
所述醚键类取代基选自以下结构:
[0019]-(ch2)
x
ch3、-(ch2ch2o)
x
ch3、-(ch2)
x
(ch2ch2o)
y
ch3、或等,其中x和y≥0 且为整数;
[0020]
所述酯键类取代基选自以下结构:
[0021]-co(ch2)
x
ch3、-co(ch2ch2o)
x
ch3、-co(ch2)
x
(ch2ch2o)
y
ch3等,其中x和y≥0 且为整数;
[0022]
所述碳酸酯键类取代基选自以下结构:
[0023]-coo(ch2)
x
ch3、-coo(ch2ch2o)
x
ch3、-coo(ch2)
x
(ch2ch2o)
y
ch3等,其中x和 y≥0且为整数;
[0024]
所述胺基甲酸酯键类取代基选自以下结构:
[0025]-conh(ch2)
x
ch3、-conh(ch2ch2o)
x
ch3、-conh(ch2)
x
(ch2ch2o)
y
ch3等,其中 x和y≥0且为整数;
[0026]
所述巯基甲酸酯键类取代基选自以下结构:
[0027]-cos(ch2)
x
ch3、-cos(ch2ch2o)
x
ch3、-cos(ch2)
x
(ch2ch2o)
y
ch3等,其中x和y ≥0且为整数;
[0028]
所述磷酸酯键类取代基选自以下结构:
[0029]-pooo(ch2)
x
ch3、-pooo(ch2ch2o)
x
ch3、-pooo(ch2)
x
(ch2ch2o)
y
ch3等,其中 x和y≥0且为整数;
[0030]
所述芳基为5~10元芳香单环或芳香稠合双环结构;
[0031]
所述杂芳基为环上含有选自o、s、n或si中的至少一种杂原子的5~10元芳香单环或芳香稠合双环结构;
[0032]
所述卤原子各自独立地选自f、cl、br、i;
[0033]
所述脂环为饱和或不饱和的3~10元单环或多环脂环;
[0034]
所述脂杂环为环上含有选自o、s、n或si中的至少一种杂原子的饱和或不饱和的 3-10元单环或多环脂杂环,所述脂杂环上含有s原子时,其任选为-s-、-so-或-so
2-;所述脂环或脂杂环上的h还可任意地被卤原子、硝基、芳基、烷基或改性烷基取代;
[0035]
所述芳环为5~10元芳香单环或芳香稠合双环;
[0036]
所述芳杂环为环上含有选自o、s、n或si中的至少一种杂原子的5~10元芳香单环或芳香稠合双环;所述芳环或芳杂环上的h还可任意地被卤原子、硝基、芳基、烷基或改性烷基取代。
[0037]
上述环状邻硝基苄基类光扳机优选选自以下环状结构:
[0038]
[0039][0040]
本发明的第二个目的是提供一系列光敏高分子衍生物。
[0041]
本发明提供的光敏高分子衍生物,包括三种结构:
[0042]
1、邻硝基苄基类光扳机修饰的光敏高分子衍生物,简称a1,具有式a
-ⅰ
结构;
[0043]
2、含双键官能团的光敏高分子衍生物,简称a2,具有式a
-ⅱ
结构;
[0044]
3、既含邻硝基苄基类光扳机又含双键官能团的光敏高分子衍生物,简称a3,具有式 a
-ⅲ
结构。
[0045][0046]
其中,邻硝基苄基类光扳机,结构如式i所示,具有两种结构,分别如结构式i-1与结构式i-2。结构式i-1表示不含环状结构的邻硝基苄基类光扳机。结构式i-2表示环状邻硝基苄基类光扳机,符号表示为cnb。
[0047][0048]
式i-1、式i-2中,x=o时,称为邻硝基苄基类光扳机,符号表示为nb,x=s时,称为邻硝基苄硫基类光扳机,符号表示为snb,x=n时,称为邻硝基苄胺基类光扳机,符号表示为nnb。
[0049]
式a
-ⅰ
、式a
-ⅲ
、式i、式i-1、式i-2中,r
’
选自氢、卤原子、羟基、巯基、胺基、硝基、氰基、醛基、酮基、酯基、酰胺基、膦酸基、膦酸酯基、磺酸基、磺酸酯基、砜基、亚砜基、芳基、杂芳基、烷基、亚烷基、改性烷基或改性亚烷基等;
[0050]
式i-1、式i-2中,r1选自氢、醚键类取代基、酯键类取代基、碳酸酯键类取代基、胺基甲酸酯键类取代基、巯基甲酸酯键类取代基或磷酸酯键类取代基等;
[0051]
式i-1、式i-2中,r2,r3,r4,r5可自由的选自氢、卤原子、羟基、巯基、胺基、硝基、氰基、醛基、酮基、羧基、酯基、酰胺基、膦酸基、膦酸酯基、磺酸基、磺酸酯基、砜基、亚砜基、芳基、杂芳基、烷基、亚烷基、改性烷基或改性亚烷基等。
[0052]
对于式i-1、式i-2所示结构,可选地,r2,r3,r4,r5可相互连接,与碳原子一起形成饱和或不饱和的脂环或脂杂环,或形成芳环或芳杂环。
[0053]
式
ⅰ-
2中,x为o、s或nh等,连接键r1一端与x连接,另一端与r2,r3,r4, r5中任意的一个基团相连接,构成环状结构。
[0054]
式a
-ⅰ
、式a
-ⅲ
中,n≥2,即单条p1高分子链上的邻硝基苄基类光扳机的平均个数大于或等于2。
[0055]
式a
-ⅰ
、式a
-ⅲ
中,p1为一种亲水或水溶性天然高聚物或合成聚合物,或p1独立的选自多种亲水或水溶性天然高聚物或合成聚合物等。
[0056]
对于含双键官能团的光敏高分子衍生物,或既含邻硝基苄基类光扳机又含双键官能团的光敏高分子衍生物而言,式a
-ⅱ
和式a
-ⅲ
中,r
’1,r
’2、r
’3选自氢、烷基、改性烷基或芳基等;r
’4选自烷基、醚键类取代基、酯键类取代基、酰胺键类取代基等;
[0057]
可选地,式a
-ⅱ
和式a
-ⅲ
中,r
’1,r
’2、r
’3可相互连接,与碳原子一起形成饱和或不饱和的脂环或脂杂环。
[0058]
式a
-ⅱ
和式a
-ⅲ
中,n≥2,即单条p1高分子链上的邻硝基苄基类光扳机的平均个数大于或等于2。p1为一种亲水或水溶性天然高聚物或合成聚合物,或p1独立的选自多种亲水或水溶性天然高聚物或合成聚合物等。
[0059]
以上三种光敏高分子衍生物中的高分子p1可以是亲水或水溶性天然高聚物,也可以是亲水或水溶性合成聚合物。
[0060]
亲水或水溶性天然高聚物包括天然多糖类物质及其修饰物或降解物,蛋白及其修饰物、改性物和降解的多肽类物质等;
[0061]
所述天然多糖类物质包括透明质酸、羧甲基纤维素、甲基纤维素、羟乙基纤维素、羟丙基纤维素、海藻酸、葡聚糖、琼脂糖、肝素、硫酸软骨素、乙二醇壳聚糖、丙二醇壳聚糖、
壳聚糖乳酸盐、羧甲基壳聚糖或壳聚糖季铵盐;
[0062]
所述蛋白包括各种亲水性或水溶性动植物蛋白、胶原蛋白、血清蛋白、丝素蛋白、弹性蛋白,所述蛋白降解物包括明胶或多肽;
[0063]
亲水或水溶性合成聚合物包括两臂或多臂聚乙二醇、聚乙烯亚胺、树枝体、合成多肽、聚赖氨酸、聚谷氨酸、聚丙烯酸、聚甲基丙烯酸、聚丙烯酸酯、聚甲基丙烯酸酯、聚丙烯酰胺、聚甲基丙烯酰胺、聚乙烯醇、聚乙烯吡咯烷酮;
[0064]
所述的以上三种光敏高分子衍生物可以是同时含有一种或一种以上不同基团的亲水或水溶性高分子,或者是一种或一种以上不同基团的亲水或水溶性高分子的混合物。
[0065]
当组分a为具有式a
-ⅰ
结构的邻硝基苄基类光扳机修饰的光敏高分子衍生物时,
[0066]
当邻硝基苄基类光扳机选择结构式i-1时,
[0067]
p1一端与r2,r3,r4,r5中任意的一个或多个基团相连接,或连接于r2,r3,r4, r5之间形成的饱和或不饱和脂环或脂杂环,或r2,r3,r4,r5之间形成的芳环或芳杂环,
[0068]
当邻硝基苄基类光扳机选择结构式i-2时,
[0069]
p1一端与r2,r3,r4,r5中任意的一个或多个基团相连接,或连接于r2,r3,r4, r5之间形成的饱和或不饱和脂环或脂杂环,或r2,r3,r4,r5之间形成的芳环或芳杂环,或连接到r1与r2,r3,r4,r5中任意的一个基团相连接构成的环状链上,
[0070]
其连接键选自羟基类所获得的连接键p
1-o-;或选自巯基类所获得的连接键p
1-s-;或选自胺基类所获得的连接键p
1-nh-;或选自烷烃类所获得的连接键p
1-;或选自酯键类所获得的连接键p
1-coo-;或选自酰胺键类所获得的连接键p
1-conh-,该连接键的一端与 p1相连,另一端连接在式a
-ⅰ
所示分子的苯环上;
[0071]
当组分a为具有式a
-ⅲ
结构的既含邻硝基苄基类光扳机又含双键官能团的光敏高分子衍生物时,
[0072]
当邻硝基苄基类光扳机选择结构式i-1时,
[0073]
p1一端与r2,r3,r4,r5中任意的一个或多个基团相连接,或连接于r2,r3,r4, r5之间形成的饱和或不饱和脂环或脂杂环,或r2,r3,r4,r5之间形成的芳环或芳杂环, p1另一端与r
’4连接;
[0074]
当邻硝基苄基类光扳机选择结构式i-2时,
[0075]
p1一端与r2,r3,r4,r5中任意的一个或多个基团相连接,或连接于r2,r3,r4, r5之间形成的饱和或不饱和脂环或脂杂环,或r2,r3,r4,r5之间形成的芳环或芳杂环,或连接到r1与r2,r3,r4,r5中任意的一个基团相连接构成的环状链上,
[0076]
p1另一端与r
’4连接;
[0077]
其连接键选自羟基类所获得的连接键-o-p
1-o-;或选自巯基类所获得的连接键-s-p
1-s-;或选自胺基类所获得的连接键-nh-p
1-nh-;或选自烷烃类所获得的连接键-p
1-;或选自酯键类所获得的连接键-coo-p
1-coo-;或选自酰胺键类所获得的连接键-conh-p
1-conh-;其连接键也可以选自在p1两端连接不同种以上所述的羟基类、巯基类、胺基类、烷烃类、酯键类、酰胺键类连接键,该连接键的一端与p1相连,另一端连接在式a
-ⅲ
所示分子的苯环上。
[0078]
其中,所述式a
-ⅰ
为邻硝基苄基类光扳机修饰的光敏高分子衍生物,所述式a
-ⅱ
为含双键官能团的光敏高分子衍生物,所述式a
-ⅲ
为既含邻硝基苄基类光扳机又含双键官能
团的光敏高分子衍生物。式a
-ⅲ
的结构设计是在式a
-ⅰ
和式a
-ⅱ
的结构基础上,在同一高分子链上同时接枝邻硝基苄基类光扳机和双键官能团,可以同时实现两种方式的交联,即同时具备光引发自由基聚合交联速度快的优势,以及光偶合反应交联组织粘附力强的优势,并且双重交联的方式,提高了水凝胶的力学性能。因此,分子结构的优化,使得其作为光敏基团修饰得到的高分子衍生物展现出更为优异的材料性能,其交联速度由单纯的醛基-胺基光偶合交联的30s左右提高到了2s以内,组织粘附力提高到80-100kpa左右,力学性能提高到1-2mpa左右,具体数据如实施例一百六十七、一百六十八、一百六十九所示。
[0079]
进一步地,所述烷基为具有1~30个碳原子的饱和或不饱和脂肪族直链或支链的烷基;
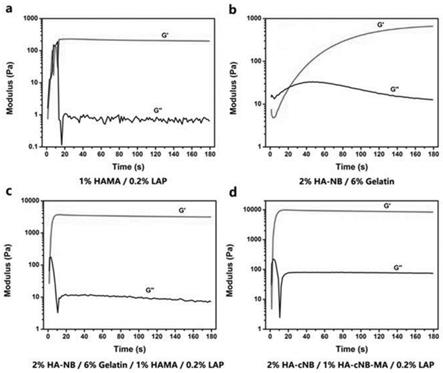
[0080]
所述亚烷基为具有1~30个碳原子的饱和或不饱和脂肪族直链或支链的亚烷基;
[0081]
所述改性烷基为烷基的任意碳原子被选自卤原子、-oh、-sh、-no2、-cn、-cho、-cooh、酯基、酰胺基、芳基、亚芳基、-co-、-o-、-s-、-so-、-so
2-、伯胺基、仲胺基、叔胺基、季铵盐基、饱和或不饱和的单环或双环亚环烃基、桥联脂杂环中的至少一种基团置换所得的基团,所述改性烷基具有1~30个原子,其碳碳单键可任意地被碳碳双键或碳碳叁键替换;
[0082]
所述改性亚烷基为亚烷基的任意碳原子被选自卤原子、-oh、-sh、-no2、-cn、-cho、
ꢀ-
cooh、酯基、酰胺基、芳基、亚芳基、-co-、-o-、-s-、-so-、-so
2-、伯胺基、仲胺基、叔胺基、季铵盐基、饱和或不饱和的单环或双环亚环烃基、桥联脂杂环中的至少一种基团置换所得的基团,所述改性亚烷基具有1~30个原子,其碳碳单键可任意地被碳碳双键或碳碳叁键替换;
[0083]
所述醚键类取代基选自以下结构:
[0084]-(ch2)
x
ch3、-(ch2ch2o)
x
ch3、-(ch2)
x
(ch2ch2o)
y
ch3、或等,其中x和y≥0 且为整数;
[0085]
所述酯键类取代基选自以下结构:
[0086]-co(ch2)
x
ch3、-co(ch2ch2o)
x
ch3、-co(ch2)
x
(ch2ch2o)
y
ch3等,其中x和y≥0 且为整数;
[0087]
所述碳酸酯键类取代基选自以下结构:
[0088]-coo(ch2)
x
ch3、-coo(ch2ch2o)
x
ch3、-coo(ch2)
x
(ch2ch2o)
y
ch3等,其中x和 y≥0且为整数;
[0089]
所述胺基甲酸酯键类取代基选自以下结构:
[0090]-conh(ch2)
x
ch3、-conh(ch2ch2o)
x
ch3、-conh(ch2)
x
(ch2ch2o)
y
ch3等,其中x和y≥0且为整数;
[0091]
所述巯基甲酸酯键类取代基选自以下结构:
[0092]-cos(ch2)
x
ch3、-cos(ch2ch2o)
x
ch3、-cos(ch2)
x
(ch2ch2o)
y
ch3等,其中x和y ≥0且为整数;
[0093]
所述磷酸酯键类取代基选自以下结构:
[0094]-pooo(ch2)
x
ch3、-pooo(ch2ch2o)
x
ch3、-pooo(ch2)
x
(ch2ch2o)
y
ch3等,其中 x和y≥0且为整数;
[0095]
所述芳基为5~10元芳香单环或芳香稠合双环结构;
[0096]
所述杂芳基为环上含有选自o、s、n或si中的至少一种杂原子的5~10元芳香单环或芳香稠合双环结构;
[0097]
所述卤原子各自独立地选自f、cl、br、i;
[0098]
所述脂环为饱和或不饱和的3~10元单环或多环脂环;
[0099]
所述脂杂环为环上含有选自o、s、n或si中的至少一种杂原子的饱和或不饱和的 3-10元单环或多环脂杂环,所述脂杂环上含有s原子时,其任选为-s-、-so-或-so
2-;所述脂环或脂杂环上的h还可任意地被卤原子、硝基、芳基、烷基或改性烷基取代;
[0100]
所述芳环为5~10元芳香单环或芳香稠合双环;
[0101]
所述芳杂环为环上含有选自o、s、n或si中的至少一种杂原子的5~10元芳香单环或芳香稠合双环;所述芳环或芳杂环上的h还可任意地被卤原子、硝基、芳基、烷基或改性烷基取代。
[0102]
进一步地,脂环或脂杂环的优选结构包括:
[0103]
等;
[0104]
进一步地,芳环或芳杂环的优选结构包括:
[0105]
等;
[0106]
r
’
的一些优选结构包括:
[0107]-h、-ch3、-ch2ch3、-ch=ch-ch=ch-ch3、-f、-cl、-br、-i、-cf3、-ccl3、-cbr3、
ꢀ-
ci3、-cn、-cooh、-ph、等。
[0108]
r2,r3,r4,r5的一些优选结构包括:
[0109]-h、-oh、-sh、-nh2、-f、-cl、-br、-i、-cf3、-ccl3、-cbr3、-ci3、-no2、-cn、
ꢀ-
cho、-cooh、-coonh2、-so3h等;
[0110]
烷基类取代基优选结构,如直链烷基-(ch2)
x
ch3、支链烷基-(ch2)
x
(cy
’
y”)
y
ch
3 (y
’
,y”为氢、烷基或改性烷基)等,其中x和y≥0且为整数;
[0111]
醚类取代基优选结构,如-o(ch2)
x
ch3、-o(ch2ch2o)
x
ch3、-o(ch2)
x
(ch2ch2o)
y
ch3等,其中x和y≥0且为整数;
[0112]
硫醚类取代基优选结构,如-s(ch2)
x
ch3、-s(ch2ch2o)
x
ch3、
ꢀ-
s(ch2)
x
(ch2ch2o)
y
ch3等,其中x和y≥0且为整数;
[0113]
胺基类取代基优选结构,如-nh(ch2)
x
ch3、-nh(ch2)
x
(cy
’
y”)
y
ch3、
ꢀ-
n(cy
’
y”)
x
(cy
’
y”)
y
、(y,y
’
为氢、烷基或改性烷基)等,其中x和y≥0且为整数;
[0114]
酯类取代基优选结构,如-coo(ch2)
x
ch3、-coo(ch2ch2o)
x
ch3、
ꢀ-
coo(ch2)
x
(ch2ch2o)
y
ch3等,其中x和y≥0且为整数;
[0115]
酰胺类取代基优选结构,如-conh(ch2)
x
ch3、-conh(ch2ch2o)
x
ch3、-conh(ch2)
x
(ch2ch2o)
y
ch3等,其中x和y≥0且为整数;
[0116]
芳香族类取代基优选结构,如-ph、等。
[0117]
邻硝基苄基类光扳机修饰的高分子衍生物中的高分子p1可以是亲水或水溶性天然高聚物包括天然多糖类物质及其修饰物或降解物,蛋白及其修饰物或降解物等,所述天然多糖类物质包括透明质酸、羧甲基纤维素、甲基纤维素、羟乙基纤维素、羟丙基纤维素、海藻酸、葡聚糖、琼脂糖、肝素、硫酸软骨素、乙二醇壳聚糖、丙二醇壳聚糖、壳聚糖乳酸盐、羧甲基壳聚糖或壳聚糖季铵盐等,所述蛋白包括各种亲水性或水溶性动植物蛋白、胶原蛋白、血清蛋白、丝素蛋白、弹性蛋白,所述蛋白降解物包括明胶或多肽等,亲水或水溶性合成聚合物包括两臂或多臂聚乙二醇、聚乙烯亚胺、树枝体、合成多肽、聚赖氨酸、聚谷氨酸、聚丙烯酸、聚甲基丙烯酸、聚丙烯酸酯、聚甲基丙烯酸酯、聚丙烯酰胺、聚甲基丙烯酰胺、聚乙烯醇、聚乙烯吡咯烷酮等。
[0118]
以上接枝或聚合的水溶或亲水性高分子衍生物中,单条高分子链上的邻硝基苄基类光扳机的平均个数大于或等于2(即n≥2)。
[0119]
所述邻硝基苄基类光扳机修饰的高分子衍生物可以是同时含有一种或一种以上不同基团的亲水或水溶性高分子,或者是一种或一种以上不同基团的亲水或水溶性高分子的混合物。所述亲水或水溶性高分子指亲水或水溶性天然高聚物,或亲水或水溶性合成聚合物。
[0120]
可选地,所述式a
-ⅰ
的邻硝基苄基类光扳机修饰的高分子衍生物,可选自以下组分 a-1~组分a-50中的结构:
[0121]
[0122]
[0123]
[0124][0125]
可选地,所述式a
-ⅰ
的邻硝基苄硫基类光扳机修饰的高分子衍生物,可选自以下组分a-51~组分a-69中的结构:
[0126]
[0127][0128]
可选地,所述式a
-ⅰ
的邻硝基苄胺基类光扳机修饰的高分子衍生物,可选自以下组分a-70~组分a-87中的结构:
[0129]
[0130][0131]
可选地,所述式a
-ⅰ
的环状邻硝基苄基类光扳机修饰的高分子衍生物,可选自以下组分a-88~组分a-106中的结构:
[0132]
[0133][0134]
组分a-1~组分a-106中,n≥2。
[0135]
可选地,所述式a
-ⅱ
的双键修饰的高分子衍生物,可选自以下组分a-107~组分a-115 中的结构:
[0136][0137]
组分a-107~组分a-115中,n≥2。
[0138]
可选地,所述式a
-ⅲ
的既含邻硝基苄基类光扳机又含双键官能团的高分子衍生物,可选自以下组分a-116~组分a-154中的结构:
[0139]
[0140]
[0141][0142]
组分a-116~组分a-154中,n≥2,ha为透明质酸;cmc为羧甲基纤维素;alg为海藻酸;cs为硫酸软骨素;pga为聚谷氨酸;peg为聚乙二醇;chitosan为壳聚糖;gelatin 为明胶;pll为聚赖氨酸;dex为葡聚糖;hep为肝素。
[0143]
结构式i-1、式i-2中,x=s时,为邻硝基苄硫基类光扳机,所示邻硝基苄硫基类光扳机修饰的高分子衍生物中,用硫原子(s)替代氧原子(o),因为硫原子的3d空轨道有利于分子内电荷转移,加快了光扳机的光解速率及光解效率,即在光照下能够更快速地、更彻底地释放出醛基/酮基或亚硝基,从而加快了其作为交联位点的交联速度,并且释放的醛基/酮基或亚硝基都能与组织表面的活性基团键连固定,能够大幅度提高材料与组织的粘附力。此外,多种活性官能团的同时释放及交联(单纯的醛基-胺基光偶合交联只是单种活性官能团的释放与交联),使得交联效率及交联密度大幅度提高,进一步提高材料的力学性能。因此,分子结构的优化,使得其作为光敏基团修饰得到的高分子衍生物展现出更为优异的材料性能,其交联速度由单纯的醛基-胺基光偶合交联的30s左右提高到了 2s以内,组织粘附力提高到80-100kpa左右,力学性能提高到1-2mpa左右,具体数据如实施例一百六十七、一百六十八、一百六十九所示。
[0144]
结构式i-1、式i-2中,x=n时,为邻硝基苄胺基类光扳机,所示邻硝基苄胺基类光扳机修饰的高分子衍生物中,用氮原子(n)替代氧原子(o),因为氮原子是强的电子给体,有利于分子内电荷转移,加快了光扳机的光解速率及光解效率,即在光照下能够更快速地、更彻底地释放出醛基/酮基或亚硝基,从而加快了其作为交联位点的交联速度,并且释放的醛基/酮基或亚硝基都能与组织表面的活性基团键连固定,能够大幅度提高材料与组织的粘附力。此外,多种活性官能团的同时释放及交联(单纯的醛基-胺基光偶合交联只是单种活
性官能团的释放与交联),使得交联效率及交联密度大幅度提高,进一步提高材料的力学性能。因此,分子结构的优化,使得其作为光敏基团修饰得到的高分子衍生物展现出更为优异的材料性能,其交联速度由单纯的醛基-胺基光偶合交联的30s左右提高到了2s以内,组织粘附力提高到80-100kpa左右,力学性能提高到1-2mpa左右,具体数据如实施例一百六十七、一百六十八、一百六十九所示。
[0145]
结构式i-2中,为环状邻硝基苄基类光扳机,具体为环状邻硝基苄基类光扳机或环状邻硝基苄硫基类光扳机或环状邻硝基苄胺基类光扳机,具有分子内环状结构,其设计目的在于光照下释放的另一活性官能团(如巯基基团等)能够保留在邻硝基苄基的母体上(单纯的醛基-胺基光偶合交联中释放的另一活性官能团会从邻硝基苄基的母体上离去),从而在同时释放醛基/酮基或亚硝基的基础上,能够额外的释放巯基,进一步增加了有效的交联位点。此外,环状邻硝基苄硫基类光扳机中,硫原子(s)的3d空轨道有利于分子内电荷转移;环状邻硝基苄胺基类光扳机中,氮原子(n)是强的电子给体,有利于分子内电荷转移,加快了光扳机的光解速率及光解效率,即在光照下能够更快速地、更彻底地释放出醛基/酮基或亚硝基,从而加快了其作为交联位点的交联速度,并且释放的醛基/酮基或亚硝基都能与组织表面的活性基团键连固定,能够大幅度提高材料与组织的粘附力。此外,多种活性官能团(醛基/酮基、亚硝基、巯基)的同时释放及交联(单纯的醛基-胺基光偶合交联只是单种活性官能团的释放与交联),使得交联效率及交联密度大幅度提高,进一步提高材料的力学性能。因此,分子结构的优化,使得其作为光敏基团修饰得到的高分子衍生物展现出更为优异的材料性能,其交联速度由单纯的醛基-胺基光偶合交联的30s 左右提高到了2s以内,组织粘附力提高到80-100kpa左右,力学性能提高到1-2mpa左右,具体数据如实施例一百六十七、一百六十八、一百六十九所示。
[0146]
本发明的第三个目的是提供所述光敏高分子衍生物的制备方法。
[0147]
2.1、提供邻硝基苄基类光扳机修饰的光敏高分子衍生物(简称a1)的制备方法。
[0148]
所述邻硝基苄基类光扳机有两种结构包括:不含环状结构的邻硝基苄基类光扳机、环状邻硝基苄基类光扳机,环状邻硝基苄基类光扳机符号表示为cnb。
[0149]
另外,所述邻硝基苄基类光扳机包括邻硝基苄基类光扳机、邻硝基苄硫基类光扳机、邻硝基苄胺基类光扳机。邻硝基苄基类光扳机符号表示为nb,邻硝基苄硫基类光扳机符号表示为snb,邻硝基苄胺基类光扳机符号表示为nnb。
[0150]
邻硝基苄基类光扳机修饰的光敏高分子衍生物(简称a1),制备方法为化学标记法和人工聚合的方法。
[0151]
其中,化学标记法是利用高分子与邻硝基苄基类光扳机中所含的化学基团间的化学反应而连接,可以是含羧基的高分子与含羟基/巯基/胺基的邻硝基苄基类小分子标记(参考文献o.p.oommen,s.wang,m.kisiel,m.sloff,j.hilborn,o.p.varghese,adv.funct. mater.2013,23,1273.);也可以是含羟基的高分子与含羧基的或含溴的邻硝基苄基类小分子标记(参考文献k.peng,i.tomatsu,a.v.korobko,a.kros,soft matter 2010,6,85;l.li, n.wang,x.jin,r.deng,s.nie,l.sun,q.wu,y.wei,c.gong,biomaterials 2014,35, 3903.);也可以是含胺基的高分子与含羧基的或含溴的邻硝基苄基类小分子标记(参考文献l.li,n.wang,x.jin,r.deng,s.nie,l.sun,q.wu,y.wei,c.gong,biomaterials 2014, 35,3903.)等标记方法。
[0152]
人工聚合的方法是利用邻硝基苄基衍生物功能单体与其它共单体共聚,可以是无规自由基聚合方法,也可以是控制自由基聚合方法(比如atrp聚合、raft聚合方法)等。
[0153]
本发明中,邻硝基苄基类光扳机修饰的高分子衍生物的一些可实施的制备方法如下:
[0154]
第一种可实施的制备方法为:将含有羧基的水溶性聚合物或高分子于蒸馏水中溶解,加入含有活性官能团羟基或巯基或胺基的邻硝基苄基小分子后,加入缩合剂1-乙基-(3
-ꢀ
二甲基胺基丙基)碳二亚胺盐酸盐(edc-hcl)和活化剂羟基苯并三唑(hobt),然后在室温下搅拌24-48h。反应结束后,将反应液加入透析袋中用稀盐酸溶液透析2-3d,然后冷冻干燥,即可得到所述的邻硝基苄基修饰的光敏高分子衍生物。
[0155]
第二种可实施的制备方法为:将含有羧基的水溶性聚合物或高分子于0.01mol/l 2-(n
-ꢀ
吗啉)乙磺酸mes缓冲溶液(ph=5.2)中,搅拌至完全溶解,将邻硝基苄基小分子溶于二甲基亚砜后加入上述反应液,将4-(4,6-二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐 (dmtmm)溶于mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应 24h。然后将反应液倒入透析袋中,用去离子水透析2-3d,然后冷冻干燥,即可得到所述的邻硝基苄基修饰的光敏高分子衍生物。
[0156]
第一种可实施方式与第二种可实施方式中,上述含有羧基的水溶性聚合物或高分子可以为聚乙二醇类、含羧基的多糖类(如:透明质酸、羧甲基纤维素、海藻酸等)、含羧基的蛋白或多肽类(如:明胶等),优选为多臂羧基聚乙二醇、透明质酸、羧甲基纤维素、明胶。进一步优选为透明质酸。
[0157]
第三种可实施的制备方法为:将含有羟基或胺基的水溶性聚合物于蒸馏水中溶解,加入含有活性官能团羧基的邻硝基苄基小分子后,加入缩合剂1-乙基-(3-二甲基胺基丙基) 碳二亚胺盐酸盐(edc-hcl)和催化剂对甲苯磺酸吡啶盐(dpts),然后在室温下搅拌 24-48h。反应结束后,将反应液倒入难溶性溶剂中重沉淀(比如修饰的聚乙二醇衍生物可倒入乙醚中重沉淀,多糖类高分子衍生物可倒入乙醇中重沉淀),然后溶于水中用透析袋透析2-3d,冷冻干燥后,即可得到所述的邻硝基苄基修饰的光敏高分子衍生物。
[0158]
第四种可实施的制备方法为:将含有羟基或胺基的水溶性聚合物于蒸馏水中溶解,加入含有活性官能团溴的邻硝基苄基小分子后,加入碳酸钾作为碱,在室温下反应24-48h。反应结束后,将反应液倒入难溶性溶剂(比如修饰的聚乙二醇衍生物可倒入乙醚中,修饰的多糖类高分子衍生物可倒入乙醇中)中重沉淀,然后溶于水中用透析袋透析2-3d,冷冻干燥后,即可得到所述的邻硝基苄基修饰的光敏高分子衍生物。
[0159]
第三种可实施方式与第四种可实施方式中,上述含有羟基或胺基的水溶性聚合物可以为含羟基或胺基的聚乙二醇类或天然多糖类或蛋白/多肽类,优选为多臂羟基聚乙二醇、多臂胺基聚乙二醇、乙二醇壳聚糖、丙二醇壳聚糖、羧甲基壳聚糖、壳聚糖乳酸盐类或天然多糖类,或聚赖氨酸、明胶等,进一步优选为乙二醇壳聚糖、多臂羟基聚乙二醇。
[0160]
上述反应中,水溶性聚合物中的羧基、羟基或胺基与小分子邻硝基苄基类衍生物的摩尔比优选为1:0.1-2;胺基修饰的邻硝基苄基类小分子与1-乙基-(3-二甲基胺基丙基)碳二亚胺盐酸盐(edc-hcl)、活化剂羟基苯并三唑(hobt)的摩尔比优选为1:2:1.5;胺基修饰的邻硝基苄基类小分子与4-(4,6-二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐(dmtmm) 的摩尔比优选为1:7.5;羧基修饰的邻硝基苄基类小分子与1-乙基-(3-二甲基胺基丙基) 碳
二亚胺盐酸盐(edc-hcl)、催化剂dpts的摩尔比优选为1:2:1.5;溴代的邻硝基苄基类小分子与碳酸钾的摩尔比优选为1:2。
[0161]
第五种可实施的制备方法为:将邻硝基苄基可聚合单体衍生物与一种或几种可聚合共单体经过聚合即可得邻硝基苄基修饰的合成共聚物。经过多次溶解-重沉淀的方法将其纯化。
[0162]
上述邻硝基苄基可聚合单体衍生物可以为丙烯酸酯类化合物、甲基丙烯酸酯类化合物、丙烯酰胺类化合物、甲基丙烯酰胺类化合物,优选为甲基丙烯酸酯类化合物和丙烯酰胺类化合物,进一步优选为甲基丙烯酸酯类化合物。
[0163]
上述可聚合共单体中至少一种必须是水溶性共单体,可以为甲基丙烯酸聚乙二醇酯 (peg-ma)、丙烯酸聚乙二醇酯、甲基丙烯酸(maa)、丙烯酸(aa)、丙烯酸羟乙酯、丙烯酰胺(am)等任意具有水溶性的可聚合单体,优选为甲基丙烯酸聚乙二醇酯 (peg-ma)。其它共单体根据不同的应用而选择。
[0164]
上述邻硝基苄基可聚合单体衍生物与水溶性共单体的聚合摩尔比可以为1:20-1:2,优选为1:9-1:3,进一步优选为1:4。
[0165]
上述聚合方法可以是无规自由基聚合、也可以是控制自由基聚合(比如raft聚合、 atrp聚合等)。优选为无规自由基聚合。即邻硝基苄基可聚合单体衍生物与共单体共溶于一定的溶剂中,加入自由基引发剂充分溶解后,经过三次冷冻-抽真空循环操作后,在加热条件下反应过夜。待反应结束后,将反应液倒入无水乙醚中沉淀,经过多次溶解-重沉淀的纯化过程,真空干燥后即可得到含邻硝基苄基的共聚合物。(参考文献g.delaittre, t.pauloehrl,m.bastmeyer,c.barner-kowollik,macromolecules 2012,45,1792-1802.)
[0166]
2.2、本发明提供含双键官能团的光敏高分子衍生物(简称a2)的制备方法。
[0167]
本发明中,双键修饰的光敏高分子衍生物的制备方法包括以下几种:
[0168]
第一种可实施的制备方法为:将含羟基或胺基的水溶性高分子溶于去离子水,冷却至 0-4℃,加入丙烯酸酐或甲基丙烯酸酐,再缓慢滴加5m naoh,反应24h,然后将反应液倒入透析袋中,用去离子水透析2-3d,然后冷冻干燥,即可得到所述的双键修饰的光敏高分子衍生物。
[0169]
上述含有羟基或胺基的水溶性聚合物或高分子可以为聚乙二醇类、含羟基或胺基的多糖类(如:透明质酸、海藻酸、羧甲基纤维素、羧甲基壳聚糖、葡聚糖、硫酸软骨素等)、含羟基或胺基的蛋白或多肽类(如:明胶等),优选为透明质酸、明胶、海藻酸、羧甲基纤维素、硫酸软骨素,进一步优选为透明质酸。
[0170]
第二种可实施的制备方法为:将含羟基或胺基的水溶性高分子溶于去离子水,加热至 40℃搅拌溶解,加入丙烯酸缩水甘油酯或甲基丙烯酸缩水甘油酯,再加入5m naoh,反应2-3h后,将反应液倒入透析袋中,用去离子水透析2-3d,然后冷冻干燥,即可得到所述的双键修饰的光敏高分子衍生物。
[0171]
上述含有羟基或胺基的水溶性聚合物或高分子可以为聚乙二醇类、含羟基或胺基的多糖类(如:透明质酸、海藻酸、羧甲基纤维素、羧甲基壳聚糖、葡聚糖、硫酸软骨素等)、含羟基或胺基的蛋白或多肽类(如:明胶等),优选为透明质酸、明胶、羧甲基壳聚糖,进一步优选为羧甲基壳聚糖。
[0172]
第三种可实施的制备方法为:将含羟基或胺基的水溶性高分子溶于无水二甲基亚
砜中,加入三乙胺,再加入丙烯酰氯或甲基丙烯酰氯(溶于二氯甲烷中),反应10h,反应结束后,将反应液倒入乙醇中重沉淀,过滤得到的粗产物重新溶于去离子水中,透析2-3d,然后冷冻干燥,即可得到所述的双键修饰的光敏高分子衍生物。
[0173]
上述含有羟基或胺基的水溶性聚合物或高分子可以为聚乙二醇类、含羟基或胺基的多糖类(如:葡聚糖等),优选为多臂聚乙二醇、葡聚糖,进一步优选为葡聚糖。
[0174]
2.3、提供既含邻硝基苄基类光扳机又含双键官能团的光敏高分子衍生物(简称a3) 的制备方法。
[0175]
所述邻硝基苄基类光扳机有两种结构包括:不含环状结构的邻硝基苄基类光扳机、环状邻硝基苄基类光扳机,环状邻硝基苄基类光扳机符号表示为cnb。
[0176]
另外,所述邻硝基苄基类光扳机包括邻硝基苄基类光扳机、邻硝基苄硫基类光扳机、邻硝基苄胺基类光扳机。邻硝基苄基类光扳机符号表示为nb,邻硝基苄硫基类光扳机符号表示为snb,邻硝基苄胺基类光扳机符号表示为nnb。
[0177]
本发明中,既含邻硝基苄基类光扳机又含双键官能团的光敏高分子衍生物(简称a3) 的制备方法为先标记邻硝基苄基类光扳机后标记双键官能团和先标记双键官能团后标记邻硝基苄基类光扳机,具体标记方法按上述邻硝基苄基类光扳机或双键官能团的标记方法制备。邻硝基苄基类光扳机标记方法是利用高分子与邻硝基苄基类光扳机中所含的化学基团间的化学反应而连接,可以是含羧基的高分子与含羟基/巯基/胺基的邻硝基苄基类小分子标记;也可以是含羟基的高分子与含羧基的或含溴的邻硝基苄基类小分子标记;也可以是含胺基的高分子与含羧基的或含溴的邻硝基苄基类小分子标记等标记方法。双键官能团标记方法是利用丙烯酸酐类分子、甲基丙烯酸酐类分子,或丙烯酸缩水甘油酯类分子、甲基丙烯酸缩水甘油酯类分子,或丙烯酰氯类分子、甲基丙烯酰氯类分子等。
[0178]
本发明中,既含邻硝基苄基类光扳机又含双键官能团的光敏高分子衍生物(简称a3) 的制备方法包括以下几种:
[0179]
第一种可实施的制备方法为:将含邻硝基苄基类光扳机的水溶性高分子溶于去离子水,冷却至0-4℃,加入丙烯酸酐或甲基丙烯酸酐,再缓慢滴加5m naoh,反应24h,然后将反应液倒入透析袋中,用去离子水透析2-3d,然后冷冻干燥,即可得到所述的既含邻硝基苄基类光扳机又含双键官能团的光敏高分子衍生物。
[0180]
第二种可实施的制备方法为:将含邻硝基苄基类光扳机的水溶性高分子溶于去离子水,加热至40℃搅拌溶解,加入丙烯酸缩水甘油酯或甲基丙烯酸缩水甘油酯,再加入5mnaoh,反应2-3h后,将反应液倒入透析袋中,用去离子水透析2-3d,然后冷冻干燥,即可得到所述的既含邻硝基苄基类光扳机又含双键官能团的光敏高分子衍生物。
[0181]
第三种可实施的制备方法为:将含邻硝基苄基类光扳机的水溶性高分子溶于无水二甲基亚砜中,加入三乙胺,再加入丙烯酰氯或甲基丙烯酰氯(溶于二氯甲烷中),反应10h,反应结束后,将反应液倒入乙醇中重沉淀,过滤得到的粗产物重新溶于去离子水中,透析 2-3d,然后冷冻干燥,即可得到所述的既含邻硝基苄基类光扳机又含双键官能团的光敏高分子衍生物。
[0182]
第四种可实施的制备方法为:将含双键官能团的水溶性聚合物或高分子于蒸馏水中溶解,加入含有活性官能团羟基或巯基或胺基的邻硝基苄基小分子后,加入缩合剂1-乙基
-ꢀ
(3-二甲基胺基丙基)碳二亚胺盐酸盐(edc-hcl)和活化剂羟基苯并三唑(hobt),然后
在室温下搅拌24-48h。反应结束后,将反应液加入透析袋中用稀盐酸溶液透析2-3d,然后冷冻干燥,即可得到所述的既含邻硝基苄基类光扳机又含双键官能团的光敏高分子衍生物。
[0183]
第五种可实施的制备方法为:将含双键官能团的水溶性聚合物或高分子于0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2)中,搅拌至完全溶解,将邻硝基苄基小分子溶于二甲基亚砜后加入上述反应液,将4-(4,6-二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐 (dmtmm)溶于mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应 24h。然后将反应液倒入透析袋中,用去离子水透析2-3d,然后冷冻干燥,即可得到所述的既含邻硝基苄基类光扳机又含双键官能团的光敏高分子衍生物。
[0184]
第六种可实施的制备方法为:将含双键官能团的水溶性聚合物溶解后,加入含有活性官能团羧基的邻硝基苄基小分子后,加入缩合剂1-乙基-(3-二甲基胺基丙基)碳二亚胺盐酸盐(edc-hcl)和催化剂对甲苯磺酸吡啶盐(dpts),然后在室温下搅拌24-48h。反应结束后,将反应液倒入难溶性溶剂中重沉淀(比如修饰的聚乙二醇衍生物可倒入乙醚中重沉淀,多糖类高分子衍生物可倒入乙醇中重沉淀),然后溶于水中用透析袋透析2-3d,冷冻干燥后,即可得到所述的既含邻硝基苄基类光扳机又含双键官能团的光敏高分子衍生物。
[0185]
第七种可实施的制备方法为:将含双键官能团的水溶性聚合物于蒸馏水中溶解,加入含有活性官能团溴的邻硝基苄基小分子后,加入碳酸钾作为碱,在室温下反应24-48h。反应结束后,将反应液倒入难溶性溶剂(比如修饰的聚乙二醇衍生物可倒入乙醚中,修饰的多糖类高分子衍生物可倒入乙醇中)中重沉淀,然后溶于水中用透析袋透析2-3d,冷冻干燥后,即可得到所述的既含邻硝基苄基类光扳机又含双键官能团的光敏高分子衍生物。
[0186]
本发明的第四个目的是提供光交联水凝胶材料的制备方法。该光交联水凝胶材料是以发明目的二所述光敏高分子衍生物为原料制备的。
[0187]
光交联水凝胶材料的制备方法,包括以下步骤:
[0188]
将组分a-光敏高分子衍生物溶于生物相容性介质得到光敏高分子溶液a;
[0189]
将组分b-光引发剂溶于生物相容性介质得到光引发剂溶液b;
[0190]
将溶液a和溶液b混合均匀得到水凝胶前体溶液,水凝胶前体溶液在光源照射下,发生光交联形成水凝胶。其交联方式为:组分a中的邻硝基苄基类光扳机和/或双键官能团和组分b-光引发剂在光照下,分别发生自由基交联(即邻硝基苄基类光扳机的自由基交联和双键官能团的自由基交联)。
[0191]
进一步的,另一种光交联水凝胶材料的制备方法,包括以下步骤:
[0192]
将组分a-光敏高分子衍生物溶于生物相容性介质得到光敏高分子溶液a;
[0193]
将组分b-光引发剂溶于生物相容性介质得到光引发剂溶液b;
[0194]
将辅助组分c-其他生物相容性高分子衍生物溶于生物相容性介质得到高分子溶液c,所述辅助组分c-其他生物相容性高分子衍生物为含胺基、联胺、酰肼或羟胺官能团的高分子衍生物,或含巯基官能团的高分子衍生物;
[0195]
将溶液a、溶液b和溶液c混合均匀得到水凝胶前体溶液,水凝胶前体溶液在光源照射下,发生光交联形成水凝胶。其交联方式为:组分a中的邻硝基苄基类光扳机和/或双键官能团和组分b-光引发剂在光照下,分别发生自由基交联(即邻硝基苄基类光扳机的自由基交联和双键官能团的自由基交联),同时组分a中的邻硝基苄基类光扳机在光照下产生的醛基/酮基与组分c中的胺基、联胺、酰肼或羟胺官能团发生光偶合交联,产生的亚硝基与组分
c中的巯基官能团发生光致亚硝基交联,是一种复合型光交联的方式。
[0196]
本发明中,水凝胶前体溶液的配置可根据不同的需要从组分a,组分b,组分c中选择,其中组分a和组分b为必要组分,组分c为辅助组分,因此水凝胶前体溶液可以为组分a/组分b,也可以为组分a/组分b/组分c。组分a的配置可以根据不同的需要从光敏高分子衍生物a1,a2,a3中选择,可以为一种或一种以上光敏高分子衍生物的混合物,但不能为单独的a2。因此,所有可能涉及的组分方式为a1/b;a3/b;a1、a2/b;a1、 a3/b;a2、a3/b;a1、a2、a3/b;a1/b/c;a3/b/c;a1、a2/b/c;a1、a3/b/c;a2、a3/b/c; a1、a2、a3/b/c。
[0197]
本发明的制备方法中,生物相容性介质选自蒸馏水、生理盐水、缓冲液和细胞培养基溶液。根据不同的应用,可选取不同的介质。
[0198]
本发明的制备方法中,混合均匀形成的水凝胶前体溶液中,若为组分a/组分b,则组分a浓度可以为0.1%wt-60%wt,优选为1%wt-10%wt,组分b的浓度可以为 0.01%wt-10%wt,优选为0.05%wt-1.0%wt,高分子总浓度可以为0.1%wt-60%wt,优选为 1%wt-10%wt;若为组分a/组分b/组分c,则组分a与组分c的质量比可以为1:0.02-50,优选为1:0.1-10,组分b的浓度可以为0.01%wt-10%wt,优选为0.05%wt-1.0%wt,高分子总浓度可以为0.1%wt-60%wt,优选为1%wt-10%wt。
[0199]
本发明的制备方法中,光源的波长根据邻硝基苄类光扳机及光引发剂的吸收波长来确定,可以为250-500nm,优选为300-450nm,进一步优选为365、375、385、395、405nm。
[0200]
本发明光交联水凝胶的制备方法采用的技术原理:分a中的邻硝基苄基类光扳机和/ 或双键官能团和组分b-光引发剂在光照下,分别发生自由基交联,同时组分a中的邻硝基苄基类光扳机在光照下产生的醛基/酮基与组分c中的胺基、联胺、酰肼或羟胺官能团发生光偶合交联,产生的亚硝基与组分c中的巯基官能团发生光致亚硝基交联,从而可以实现在一次光照下的多重交联,是一种复合型光交联的方式。
[0201]
对于光交联水凝胶材料的制备方法中,
[0202]
组分b-光引发剂,即光照下能够产生自由基的物质,优选为水溶性光引发剂或可分散在水中的光引发剂,进一步优选为i 2959(组分b-1),lap(组分b-2),eosin-y(组分b-3)等及其衍生物。
[0203][0204]
组分c-含胺基、联胺、酰肼或羟胺官能团的高分子衍生物,分别具有结构式c
-ⅰ
、 c
-ⅱ
、c
-ⅲ
、c
-ⅳ
;含巯基官能团的高分子衍生物,具有结构式c
-ⅴ
:
[0205][0206]
结构式c
-ⅰ
、c
-ⅱ
、c
-ⅲ
、c
-ⅳ
、c
-ⅴ
中,n≥2,p2、p3、p4、p5、p6为亲水或水溶性天然高
聚物,也可以是亲水或水溶性合成聚合物等。
[0207]
亲水或水溶性天然高聚物包括天然多糖类物质及其修饰物或降解物,蛋白及其修饰物、改性物和降解的多肽类物质等。
[0208]
所述天然多糖类物质包括透明质酸、羧甲基纤维素、甲基纤维素、羟乙基纤维素、羟丙基纤维素、海藻酸、葡聚糖、琼脂糖、肝素、硫酸软骨素、乙二醇壳聚糖、丙二醇壳聚糖、壳聚糖乳酸盐、羧甲基壳聚糖或壳聚糖季铵盐。
[0209]
所述蛋白包括各种亲水性或水溶性动植物蛋白、胶原蛋白、血清蛋白、丝素蛋白、弹性蛋白,所述蛋白降解物包括明胶或多肽。
[0210]
亲水或水溶性合成聚合物包括两臂或多臂聚乙二醇、聚乙烯亚胺、树枝体、合成多肽、聚赖氨酸、聚谷氨酸、聚(甲基)丙烯酸、聚(甲基)丙烯酸酯、聚(甲基)丙烯酰胺、聚乙烯醇、聚乙烯吡咯烷酮。
[0211]
含胺基、联胺、酰肼、羟胺或巯基基团的高分子衍生物可以是同时含有一种或一种以上不同基团的亲水或水溶性天然高聚物或合成聚合物,或者是一种或一种以上不同基团的亲水或水溶性天然高聚物或合成聚合物。
[0212]
对于含胺基、联胺、酰肼、羟胺等高分子衍生物,式c
-ⅰ
所示结构,代表含n个胺基基团的水溶性或亲水性的高分子。式c
-ⅱ
所示结构,代表含n个联胺基基团的水溶性或亲水性的高分子。式c
-ⅲ
所示结构,代表含n个酰肼基基团的水溶性或亲水性的高分子。式c
-ⅳ
所示结构,代表含n个羟胺基基团的水溶性或亲水性的高分子。式c
-ⅴ
所示结构,代表含n个巯基基团的水溶性或亲水性的高分子。
[0213]
可选地,所述式c
-ⅰ
可选自以下组分c-1~组分c-9中的结构;所述式c
-ⅱ
可选自以下组分c-10中的结构;所述式c
-ⅲ
可选自以下组分c-11~组分c-13中的结构;所述式 c
-ⅳ
可选自以下组分c-14~组分c-15中的结构;所述式c
-ⅴ
选自以下组分c-16~组分c-21 中的结构:
[0214]
[0215][0216]
组分c-1~组分c-21中,n≥2,组分c-1为壳聚糖;组分c-2为乙二醇壳聚糖;组分 c-3为羧甲基壳聚糖;组分c-4为明胶;组分c-5为聚赖氨酸;组分c-6为聚乙烯亚胺;组分c-7为两臂胺基聚乙二醇;组分c-8为四臂胺基聚乙二醇;组分c-9为胺基聚合物;组分c-10为联胺修饰的羧甲基纤维素;组分c-11~组分c-13为酰肼修饰的透明质酸;组分c-14为四臂羟胺聚乙二醇;组分c-15为羟胺修饰的葡聚糖;组分c-16为两臂巯基聚乙二醇;组分c-17为四
臂巯基聚乙二醇;组分c-18为巯基修饰的透明质酸;组分c-19 为巯基修饰的壳聚糖;组分c-20为巯基修饰的葡聚糖;组分c-21为巯基修饰的肝素。
[0217]
本发明还提供组分c-含胺基、联胺、酰肼或羟胺基团的高分子衍生物的制备方法。
[0218]
本发明中,胺基修饰的水溶性聚合物可以是人工合成聚胺类高分子及其修饰物(如聚乙烯亚胺pei、树枝体pamam,两臂或多臂胺基聚乙二醇),或天然含胺基多糖类亲水或水溶性高分子及其修饰物或降解物(如乙二醇壳聚糖、丙二醇壳聚糖、壳聚糖乳酸盐、羧甲基壳聚糖、壳寡糖等);也可以是生物或经微生物表达后提取的蛋白及其改性物或降解物(如胶原,血清蛋白及明胶等);也可以是人工合成或通过微生物表达并提取的含两个或两个胺基以上的亲水或水溶性多肽(如聚赖氨酸等)或丙烯酸酯或甲基丙烯酸酯或丙烯酰胺类或甲基丙烯酰胺类聚合物及其修饰物。优选为明胶、乙二醇壳聚糖。
[0219]
本发明中,联胺修饰的高分子衍生物的制备方法为:将含有羧基的水溶性聚合物和二联胺于蒸馏水中溶解,加入缩合剂1-乙基-(3-二甲基胺基丙基)碳二亚胺盐酸盐(edc-hcl) 和活化剂羟基苯并三唑(hobt),然后在室温下搅拌24-48h。反应结束后,将反应液倒入透析袋中用稀盐酸溶液透析2-3d,然后冷冻干燥,即可得到所述的联胺修饰的高分子衍生物。
[0220]
上述含有羧基的水溶性聚合物可以为羧基聚乙二醇类、含羧基的多糖类(如壳聚糖乳酸盐、羧甲基壳聚糖、透明质酸、海藻酸、羧甲基纤维素等),优选为多臂羧基聚乙二醇、透明质酸,进一步优选为透明质酸。
[0221]
上述反应中,水溶性聚合物中的羧基与小分子二联胺的摩尔比优选为1:0.1-2;二联胺小分子与1-乙基-(3-二甲基胺基丙基)碳二亚胺盐酸盐(edc-hcl)、活化剂羟基苯并三唑(hobt)的摩尔比优选为1:2:1.5。
[0222]
本发明中,酰肼修饰的高分子衍生物的制备方法为:将含有羧基的水溶性聚合物和二酰肼于蒸馏水中溶解,加入缩合剂1-乙基-(3-二甲基胺基丙基)碳二亚胺盐酸盐(edc-hcl) 和活化剂羟基苯并三唑(hobt),然后在室温下搅拌24-48h。反应结束后,将反应液倒入透析袋中用稀盐酸溶液透析2-3d,然后冷冻干燥,即可得到所述的酰肼修饰的高分子衍生物。
[0223]
上述含有羧基的水溶性聚合物可以为羧基聚乙二醇类、含羧基的多糖类(如壳聚糖乳酸盐、羧甲基壳聚糖、透明质酸、海藻酸、羧甲基纤维素等),优选为多臂羧基聚乙二醇、透明质酸,进一步优选为透明质酸。
[0224]
上述反应中,小分子二酰肼可以为碳二酰肼、草酸二酰肼、丙二酸二酰肼、丁二酸二酰肼、戊二酸二酰肼、己二酸二酰肼、庚二酸二酰肼等任意二酰肼,优选为碳二酰肼、草酸二酰肼、己二酸二酰肼,进一步优选为碳二酰肼。水溶性聚合物中的羧基与小分子二酰肼的摩尔比优选为1:0.1-2;二酰肼小分子与1-乙基-(3-二甲基胺基丙基)碳二亚胺盐酸盐(edc-hcl)、活化剂羟基苯并三唑(hobt)的摩尔比优选为1:2:1.5。
[0225]
本发明中,羟胺修饰的高分子衍生物的制备方法为:将含有羟基的聚合物与n-羟基邻苯二甲酰亚胺溶于二氯甲烷溶液中,加入三苯基膦后,慢慢滴加二异丙基偶氮二羧酸酯并反应16-24h后,将聚合物在乙醚中沉淀出来,然后重新溶到二氯甲烷溶液中,加入水合肼反应1-3h后,即可得到羟胺修饰的高分子衍生物。
[0226]
上述含有羟基的聚合物可以为聚乙二醇类,多糖类(如葡聚糖、壳聚糖),优选为多
臂羟基聚乙二醇。
[0227]
上述反应中,聚合物中的羟基与n-羟基邻苯二甲酰亚胺、三苯基膦、二异丙基偶氮二羧酸酯、水合肼的摩尔比优选为1:10:10:10:10。
[0228]
本发明还提供组分c-含巯基基团的高分子衍生物的制备方法。
[0229]
含巯基类基团的高分子衍生物,即巯基修饰的高分子衍生物的制备方法为化学标记法,具体是利用高分子与含巯基的衍生物中所含化学基团间的化学反应而连接,可以是含羧基的高分子与含胺基或含酰肼或含羟胺的小分子标记(参考文献amy fu,kihak gwon,juliaa.kornfield,biomacromolecules.2015,16,497.;tugba ozdemir,swati pradhan-bhatt, xinqiao jia,acs biomater.sci.eng.2016,2,2217.),也可以是含羟基的高分子与含羧基或含溴的小分子标记(参考文献rayun choi,yong-min huh,seungjoo haam,langmuir.2010, 26,17520.),也可以是含胺基的高分子与含羧基或含溴的小分子标记(参考文献hanweizhang,aisha qadeer,weiliam chen,biomacromolecules.2011,12,1428.)等标记方法。
[0230]
巯基修饰的高分子衍生物的制备方法包括以下几种:
[0231]
第一种可实施的制备方法为:将含有羧基的水溶性聚合物或高分子于蒸馏水中溶解,加入含有活性官能团胺基或酰肼或羟胺的带巯基的小分子后,加入缩合剂1-乙基-(3-二甲基胺基丙基)碳二亚胺盐酸盐(edc-hcl)和活化剂羟基苯并三唑(hobt),然后在室温下搅拌24-48h。反应结束后,将反应液加入透析袋中用稀盐酸溶液透析2-3d,然后冷冻干燥,即可得到所述的巯基修饰的高分子衍生物。
[0232]
上述含有羧基的水溶性聚合物或高分子可以为聚乙二醇类、含羧基的多糖类(如:透明质酸、羧甲基纤维素、海藻酸、肝素等),优选为多臂羧基聚乙二醇、透明质酸、肝素,进一步优选为透明质酸、肝素。
[0233]
第二种可实施的制备方法为:将含有羟基或胺基的水溶性聚合物或高分子于蒸馏水中溶解,加入含有活性官能团羧基的带巯基的小分子后,加入缩合剂1-乙基-(3-二甲基胺基丙基)碳二亚胺盐酸盐(edc-hcl)和催化剂4-(二甲胺基)吡啶,然后在室温下搅拌24-48h。反应结束后,将反应液倒入难溶性溶剂中重沉淀(比如修饰的聚乙二醇衍生物可倒入乙醚中重沉淀,多糖类高分子衍生物可倒入乙醇中重沉淀),然后溶于水中用透析袋透析2-3d,冷冻干燥后,即可得到所述的巯基修饰的高分子衍生物。
[0234]
上述含有羟基的水溶性聚合物或高分子可以为聚乙二醇类或天然多糖类,优选为多臂聚乙二醇、葡聚糖,进一步优选为葡聚糖。上述含有胺基的水溶性聚合物或高分子可以为聚乙二醇类或天然多糖类或蛋白及多肽类,优选为多臂胺基聚乙二醇、乙二醇壳聚糖、丙二醇壳聚糖、羧甲基壳聚糖、壳聚糖乳酸盐类或蛋白及多肽类,进一步优选为羧甲基壳聚糖。
[0235]
第三种可实施的制备方法为:将含有羟基或胺基的水溶性聚合物或高分子于蒸馏水中溶解,加入含有活性官能团溴的带巯基保护基的小分子后,加入碳酸钾作为碱,在室温下反应24-48h。反应结束后,将反应液倒入难溶性溶剂(比如修饰的聚乙二醇衍生物可倒入乙醚中,修饰的多糖类高分子衍生物可倒入乙醇中)中重沉淀,然后将粗产物溶于蒸馏水中,加入dtt脱保护,反应一段时间后,将反应液倒入透析袋透析2-3d,冷冻干燥后,即可得到所述的巯基修饰的高分子衍生物。
[0236]
上述含有羟基的水溶性聚合物或高分子可以为聚乙二醇类或天然多糖类,优选为多臂聚乙二醇、葡聚糖,进一步优选为葡聚糖。上述含有胺基的水溶性聚合物或高分子可以为聚乙二醇类或天然多糖类或蛋白及多肽类,优选为多臂胺基聚乙二醇、乙二醇壳聚糖、丙二醇壳聚糖、羧甲基壳聚糖、壳聚糖乳酸盐类或蛋白及多肽类,进一步优选为羧甲基壳聚糖。
[0237]
上述反应中,水溶性高分子中的羧基、羟基或胺基与小分子巯基类衍生物的摩尔比优选为1:0.1-2;胺基或酰肼或羟胺修饰的含巯基类小分子与1-乙基-(3-二甲基胺基丙基) 碳二亚胺盐酸盐(edc-hcl)、活化剂羟基苯并三唑(hobt)的摩尔比优选为1:1.5:1.5,羧基修饰的含巯基类小分子与1-乙基-(3-二甲基胺基丙基)碳二亚胺盐酸盐(edc-hcl)、催化剂4-(二甲胺基)吡啶的摩尔比优选为1:1.5:1.5,溴代的含巯基类小分子与碳酸钾的摩尔比优选为1:2。
[0238]
本发明的第五个目的是提供利用发明目的四所述光交联水凝胶材料制备方法制得的产品,即光交联水凝胶材料,也可以称为复合型光交联水凝胶材料。
[0239]
本发明的第六个目的是提供用于本发明方法制备水凝胶的试剂盒。
[0240]
第一种试剂盒,包含:组分a-光敏高分子衍生物;组分b-光引发剂,及有关水凝胶制备及应用的说明书;
[0241]
这里组分a-光敏高分子衍生物包括两种结构:
[0242]
1、邻硝基苄基类光扳机修饰的光敏高分子衍生物,简称a1,具有上文式a
-ⅰ
结构;
[0243]
2、既含邻硝基苄基类光扳机又含双键官能团的光敏高分子衍生物,简称a3,具有上文式a
-ⅲ
结构。
[0244]
其中,邻硝基苄基类光扳机,结构如上文式i所示,具有两种结构,分别如上文结构式i-1与上文结构式i-2。结构式i-1表示不含环状结构的邻硝基苄基类光扳机。结构式i-2 表示环状邻硝基苄基类光扳机,符号表示为cnb。式i-1或式i-2中,x=o时,称为邻硝基苄基类光扳机,符号表示为nb,x=s时,称为邻硝基苄硫基类光扳机,符号表示为snb,x=n时,称为邻硝基苄胺基类光扳机,符号表示为nnb。
[0245]
组分b-光引发剂,即光照下能够产生自由基的物质,优选为水溶性光引发剂或可分散在水中的光引发剂,进一步优选为i 2959(组分b-1),lap(组分b-2),eosin-y(组分b-3)等及其衍生物。
[0246]
第二种试剂盒,除第一种试剂盒内容以外,在第一种试剂盒所述组分a-光敏高分子衍生物中还加入含双键官能团的光敏高分子衍生物,含双键官能团的光敏高分子衍生物,简称a2,具有上文式a
-ⅱ
结构。
[0247]
第三种试剂盒,在第一种试剂盒的基础上,还包括辅助组分c,所述辅助组分c为其他生物相容性高分子衍生物,包括含胺基、联胺、酰肼、羟胺或巯基官能团的高分子衍生物。辅助组分c的定义与发明目的四所述光交联水凝胶材料制备方法中辅助组分c的定义相同。
[0248]
第四种试剂盒,在第二种试剂盒的基础上,还包括辅助组分c,所述辅助组分c为其他生物相容性高分子衍生物,包括含胺基、联胺、酰肼、羟胺或巯基官能团的高分子衍生物。辅助组分c的定义与发明目的四所述光交联水凝胶材料制备方法中辅助组分c的定义相同。
[0249]
对于以上四种试剂盒中还可包含生物相容性介质,如蒸馏水、生理盐水、缓冲液和细胞培养基。
[0250]
对于以上四种试剂盒中的说明书上记载着水凝胶的应用包括其在术后创面封闭、组织液渗漏封堵、止血材料、组织工程支架材料、3d打印的生物墨水及作为细胞、蛋白或药物载体上的应用。
[0251]
本发明的第七个目的是提供光交联水凝胶材料制备方法所制得产品,即光交联水凝胶的应用。
[0252]
本发明提供了上述光交联水凝胶作为制备术后创面封闭-皮肤修复材料或药物的应用。
[0253]
本发明还提供了上述光交联水凝胶作为制备术后创面封闭-术后防黏连材料或药物的应用。
[0254]
本发明还提供了上述光交联水凝胶作为制备术后创面封闭-口腔溃疡材料或药物的应用。
[0255]
本发明还提供了上述光交联水凝胶作为制备组织液渗漏封堵-肠漏封堵材料或药物的应用。
[0256]
本发明还提供了上述光交联水凝胶作为制备组织液渗漏封堵-手术缝合材料或药物的应用。
[0257]
本发明还提供了上述光交联水凝胶作为制备止血材料-肝脏止血材料或药物的应用。
[0258]
本发明还提供了上述光交联水凝胶作为制备止血材料-骨断面止血材料或药物的应用。
[0259]
本发明还提供了上述光交联水凝胶作为制备止血材料-动脉止血材料或药物的应用。
[0260]
本发明还提供了上述光交联水凝胶作为制备止血材料-心脏止血材料或药物的应用。
[0261]
本发明还提供了上述光交联水凝胶作为制备组织工程支架材料-软骨修复材料或药物的应用。
[0262]
本发明还提供了上述光交联水凝胶作为制备组织工程支架材料-骨修复材料或药物的应用。
[0263]
本发明还提供了上述光交联水凝胶作为制备组织工程支架材料-骨/软骨复合缺损修复材料或药物的应用。
[0264]
本发明还提供了上述光交联水凝胶在3d打印(fdm)材料-生物墨水的应用。
[0265]
本发明还提供了上述光交联水凝胶在3d打印(dlp)材料-生物墨水的应用。
[0266]
本发明还提供了上述光交联水凝胶作为制备细胞、蛋白、药物载体上的应用。
[0267]
本发明中,所述式a
-ⅰ
为邻硝基苄基类光扳机修饰的光敏高分子衍生物,所述式a
-ⅱ
为含双键官能团的光敏高分子衍生物,所述式a
-ⅲ
为既含邻硝基苄基类光扳机又含双键官能团的光敏高分子衍生物。由式a
-ⅰ
、式a
-ⅱ
、式a
-ⅲ
中的一种或多种光敏高分子衍生物构成组分a;将组分a-光敏高分子衍生物溶于生物相容性介质得到光敏高分子溶液 a;将组分b-光引发剂溶于生物相容性介质得到光引发剂溶液b;将辅助组分c-其他生物相容性高分子衍生物溶于生物相容性介质得到高分子溶液c。将溶液a和溶液b(或加入溶液c)混合均匀得到水凝胶前体溶液,水凝胶前体溶液在光源照射下,发生光交联形成水凝胶。其交联方
式可以分为以下两种:
[0268]
方式一:将溶液a和溶液b混合均匀得到水凝胶前体溶液,水凝胶前体溶液在光源照射下,发生光交联形成水凝胶。其交联方式为:组分a中的邻硝基苄基类光扳机和/或双键官能团和组分b-光引发剂在光照下,分别发生自由基交联(即邻硝基苄基类光扳机的自由基交联和双键官能团的自由基交联)。邻硝基苄基类光扳机的自由基交联是通过邻硝基苄基在光照下产生的亚硝基捕获光引发剂在光照下产生的自由基,形成活性极强的亚硝基自由基,产生的亚硝基自由基可以自身发生二聚交联,也可以与组分a中的其他活性基团(如巯基、羟基、胺基、羧基、磺酸基、羰基、双键等)进行加成交联形成水凝胶。由于亚硝基自由基的反应活性比单纯亚硝基的活性更高,可以进一步提高水凝胶的交联速度及交联效率。双键官能团的自由基交联是通过光引发剂在光照下产生的自由基转移到双键上,进而引发双键的聚合交联。以上两种自由基交联方式可以只进行一种交联,即组分 a单独选用式a
-ⅰ
或式a
-ⅱ
所示的光敏高分子衍生物;也可以在一次光照下同时进行,即组分a单独选用式a
-ⅲ
所述的光敏高分子衍生物,或同时选用式a
-ⅰ
、式a
-ⅱ
、式 a
-ⅲ
中两种以上光敏高分子衍生物。该类光交联方式可以同时具备光引发自由基聚合交联速度快的优势,以及邻硝基苄基类光扳机交联组织粘附力强的优势,并且多重交联的方式,可以进一步提高水凝胶的力学性能。因此,其交联速度由单纯的醛基-胺基光偶合交联的30s左右提高到了2s以内,组织粘附力提高到80-100kpa左右,力学性能提高到1-2 mpa左右,具体数据如实施例一百六十七、一百六十八、一百六十九所示。
[0269]
方式二:将溶液a、溶液b和溶液c混合均匀得到水凝胶前体溶液,水凝胶前体溶液在光源照射下,发生光交联形成水凝胶。其交联方式为:组分a中的邻硝基苄基类光扳机和/或双键官能团和组分b-光引发剂在光照下,分别发生自由基交联(即邻硝基苄基类光扳机的自由基交联和双键官能团的自由基交联)。邻硝基苄基类光扳机的自由基交联是通过邻硝基苄基在光照下产生的亚硝基捕获光引发剂在光照下产生的自由基,形成活性极强的亚硝基自由基,产生的亚硝基自由基可以自身发生二聚交联,也可以与组分a中的其他活性基团(如巯基、羟基、胺基、羧基、磺酸基、羰基、双键等)进行加成交联形成水凝胶。由于亚硝基自由基的反应活性比单纯亚硝基的活性更高,可以进一步提高水凝胶的交联速度及交联效率。双键官能团的自由基交联是通过光引发剂在光照下产生的自由基转移到双键上,进而引发双键的聚合交联。同时组分a中的邻硝基苄基类光扳机在光照下产生的醛基/酮基与组分c中的胺基、联胺、酰肼或羟胺官能团发生席夫碱交联,产生的亚硝基与组分c中的巯基官能团发生光致亚硝基交联。以上两种自由基交联方式可以只进行一种交联,即组分a单独选用式a
-ⅰ
或式a
-ⅱ
所示的光敏高分子衍生物;也可以在一次光照下同时进行,即组分a单独选用式a
-ⅲ
所述的光敏高分子衍生物,或同时选用式a
-ⅰ
、式a
-ⅱ
、式a
-ⅲ
中两种以上光敏高分子衍生物。该类光交联方式可以同时具备光引发自由基聚合交联速度快的优势,以及邻硝基苄基类光扳机交联组织粘附力强的优势,并且多重交联的方式,可以进一步提高水凝胶的力学性能。因此,其交联速度由单纯的醛基-胺基光偶合交联的30s左右提高到了2s以内,组织粘附力提高到80-100kpa左右,力学性能提高到1-2mpa左右,具体数据如实施例一百六十七、一百六十八、一百六十九所示。
[0270]
下式为该类光交联水凝胶的交联示意式:
[0271]
1.邻硝基苄基类光扳机的自由基交联
[0272][0273]
2.双键官能团的自由基交联
[0274][0275]
本发明中,该类光交联水凝胶是在现有光交联方式(即单纯的醛基-胺基光偶合交联或光引发自由基聚合交联)的基础上,提出的一种新型光交联凝胶技术。组分b-光引发剂的引入,不仅提高了原有邻硝基苄基类光扳机的交联速度及交联效率(通过产生极强反应活性的亚硝基自由基进行交联),而且可以将光引发自由基聚合交联的高分子衍生物和光偶合反应交联的高分子衍生物混合后形成复合的光敏高分子溶液,在一次光照下,可以激活引发剂产生自由基,分别进行自由基交联(即邻硝基苄基类光扳机的自由基交联和双键官能团的自由基交联),也可以发生光偶合反应交联(即邻硝基苄基类光扳机在光照下产生的醛基/酮基与组分c中的胺基、联胺、酰肼或羟胺官能团发生席夫碱交联)及光致亚硝基交联(即邻硝基苄基类光扳机在光照下产生的亚硝基与组分c中的巯基官能团发生光致亚硝基交联),实现多重光交联制备复合型水凝胶。
[0276]
本发明与现有技术相比具有下列创新点:
[0277]
(1)光固化速度快,能在1-2s达到成胶点,10-20s达到最终模量,由于一次实现了多重光交联,其光固化速度优于单纯的光引发自由基聚合交联和光偶合交联;
[0278]
(2)组织粘附力强,能在组织表面原位凝胶化,同时光照产生的醛基/酮基和亚硝基能与组织表面的巯基、胺基、羧基发生反应,实现水凝胶与周围组织的化学键键连一体化整合,克服了自由基聚合交联需要额外底涂的问题;
[0279]
(3)力学性能优异,具有较好的延展性和强度,克服了当前大多数水凝胶机械性能差、柔弱易碎的问题;
[0280]
(4)生物相容性好,原料主要来源于天然高分子材料,且形成的水凝胶可降解;
[0281]
(5)临床操作便捷,由于光交联具有优异的时空可控性,使用时可将水凝胶前体溶液涂抹或喷涂于创面组织,在光照下快速成胶同步与组织整合,无需底涂,一步实现创面封闭;
[0282]
(6)凝胶的化学结构、组成和降解性以及强度、厚薄可调,可以根据不同的应用而灵活地调节凝胶材料的组成和性质,尤其可以在创面原位成薄胶,特别适用于术后创面的封闭和修复,也适用于组织液渗漏封堵,同时可作为止血材料,也可作为组织工程支架材料,也可以用于3d打印的生物墨水,还可以为细胞、蛋白或药物提供一种原位载体,有效应用于再生医学。
[0283]
因此,该光交联水凝胶体系的技术突破,有望实质性推动光原位凝胶技术的临床应用。
附图说明
[0284]
注:nb为本发明组分a-1中的邻硝基苄基类光扳机;cnb为本发明组分a-88中的环状邻硝基苄基类光扳机;cnb-ma为本发明组分a-144中的既含环状邻硝基苄基类光扳机又含双键官能团。其中,ha-nb即为组分a-1;ha-cnb即为组分a-88;ha-cnb-ma 即为组分a-144。
[0285]
图1为水凝胶前体溶液(2%ha-nb/6%gelatin/1%hama/0.2%lap或2%ha-cnb/1% ha-cnb-ma/0.2%lap)光照成胶的实时流变图。
[0286]
图2为该水凝胶(2%ha-nb/6%gelatin/1%hama/0.2%lap或2%ha-cnb/1% ha-cnb-ma/0.2%lap)的粘附力测试图。
[0287]
图3为该水凝胶(2%ha-nb/6%gelatin/1%hama/0.2%lap或2%ha-cnb/1% ha-cnb-ma/0.2%lap)的压缩测试图。
[0288]
图4为该水凝胶(ha-nb/gelatin/hama/lap或ha-cnb/ha-cnb-ma/lap) 的生物相容性测试图。
[0289]
图5为该水凝胶(组分a-1/组分a-107/组分c-4/组分b-2)的创面封闭的效果直观图。
[0290]
图6为该水凝胶(组分a-1/组分a-107/组分c-4/组分b-2)作为术后防黏连的效果直观图。
[0291]
图7为该水凝胶(组分a-1/组分a-107/组分c-4/组分b-2)作为肝脏止血的效果直观图。
[0292]
图8为该水凝胶(组分a-1/组分a-107/组分c-4/组分b-2)作为骨/软骨组织工程支架材料的效果直观图。
[0293]
图9为该水凝胶(组分a-1/组分a-107/组分c-4/组分b-2)作为生物墨水的打印效果直观图。
具体实施方式
[0294]
以下用实施例对本发明作更详细的描述。下面结合附图以及实施例对本发明作进一步描述,但这些实施例仅仅是对本发明最佳实施方式的描述,并不对本发明的范围有任何限制。本领域技术人员在不背离本发明精神和保护范围的情况下作出的其它任何变化和修改,仍包括在本发明保护范围之内。
[0295]
实施例一:组分a-1的合成
[0296][0297]
(1)化合物1的合成:按参考文献yunlong yang;jieyuan zhang;zhenzhen liu;
qiuning
ꢀꢀ
lin;xiaolin liu;chunyan bao;yang wang;linyong zhu.adv.mater.2016,28,2724.公开的方法进行合成。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.96(s,2h),4.13(t, j=6.1hz,2h),3.99(s,3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j =7.2hz,2h),2.26-2.17(m,2h).ms(esi):[m+h]328.1507.
[0298]
(2)组分a-1的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 1(65mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6-二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋 (mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物a-1 (1.85g),根据核磁氢谱图,可计算出化合物1的标记率大约为3.42%。
[0299]
实施例二:组分a-2的合成
[0300][0301]
(1)化合物2的合成:按参考文献james f.cameron.;jean m.j.frechet.j.am.chem. soc.1991,113,4303.公开的方法进行合成。
[0302]
(2)化合物3的合成:将化合物2(1g,3.2mmol)和乙二胺(1.1ml)溶于甲醇 (50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物3(0.89g,产率82%)。1h nmr (400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.96(m,1h),4.13(t,j=6.1hz,2h),3.99(s, 3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26
-ꢀ
2.17(m,2h),1.33(d,j=6.9hz,3h).ms(esi):[m+h]342.1624.
[0303]
(3)组分a-2的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 3(68mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6-二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋 (mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物a-2 (1.92g),根据核磁氢谱图,可计算出化合物3的标记率大约为3.29%。
[0304]
实施例三:组分a-3的合成
[0305]
[0306]
(1)化合物4的合成:按参考文献michael c.pirrung.;yong rok lee.;kaapjoo.;jamesb.springer.j.org.chem.1999,64,5042.公开的方法进行合成。
[0307]
(2)化合物5的合成:将化合物4(1g,2.7mmol)和乙二胺(1.1ml)溶于甲醇 (50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物5(0.80g,产率74%)。1h nmr (400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),6.35(dd,j=10.0,15.0hz,1h),6.04(m, 1h),5.8(m,1h),5.4(m,1h),4.96(m,1h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.32(dd,j= 11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h),1.75(d, j=6.5hz,3h).ms(esi):[m+h]394.1908.
[0308]
(3)组分a-3的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 5(79mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6-二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋 (mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物a-3 (1.73g),根据核磁氢谱图,可计算出化合物5的标记率大约为2.97%。
[0309]
实施例四:组分a-4的合成
[0310][0311]
(1)化合物6的合成:按参考文献isabelle aujard.;chouaha benbrahim.;ludovicjullien.chem.eur.j.2006,12,6865.公开的方法进行合成。
[0312]
(2)化合物7的合成:将化合物6(1g,3.1mmol)和乙二胺(1.1ml)溶于甲醇 (50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物7(0.85g,产率78%)。1h nmr (400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.96(s,1h),4.13(t,j=6.1hz,2h),3.99(s, 3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26
-ꢀ
2.17(m,2h).ms(esi):[m+h]353.1426.
[0313]
(3)组分a-4的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 7(70mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6-二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋 (mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物a-4 (1.78g),根据核磁氢谱图,可计算出化合物7的标记率大约为2.49%。
[0314]
实施例五:组分a-5的合成
[0315][0316]
(1)化合物8的合成:按参考文献alexander g.russell.;dario m.bassani.;john s. snaith.j.org.chem.2010,75,4648.公开的方法进行合成。
[0317]
(2)化合物9的合成:将化合物8(1g,2.9mmol)和乙二胺(1.1ml)溶于甲醇 (50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物9(0.78g,产率72%)。1h nmr (400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.96(s,1h),4.13(t,j=6.1hz,2h),3.99(s, 3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26
-ꢀ
2.17(m,2h).ms(esi):[m+h]372.1424.
[0318]
(3)组分a-5的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 9(74mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6-二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋 (mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物a-5 (1.76g),根据核磁氢谱图,可计算出化合物9的标记率大约为3.08%。
[0319]
实施例六:组分a-6的合成
[0320][0321]
(1)化合物10的合成:按参考文献alexandre specht.;maurice goeldner.angew.chem. int.ed.2004,43,2008.公开的方法进行合成。
[0322]
(2)化合物11的合成:将化合物10(1g,2.7mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物11(0.68g,产率63%)。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.96(s,1h),4.13(t,j=6.1hz, 2h),3.99(s,3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz, 2h),2.26-2.17(m,2h).ms(esi):[m+h]396.1374.
[0323]
(3)组分a-6的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 11(79mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔
1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-6(1.79g),根据核磁氢谱图,可计算出化合物11的标记率大约为2.34%。
[0324]
实施例七:组分a-7的合成
[0325][0326]
(1)化合物12的合成:按参考文献jack e.baldwin.;adrian w.mcconnaughie.;sungbo shin.tetrahedron.1990,46,6879.公开的方法进行合成。
[0327]
(2)化合物13的合成:将化合物12(1g,2.4mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物13(0.61g,产率57%)。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.75(ddd,j=8.2,1.4,0.4hz,1h),7.22(s,1h), 7.57(tdd,j=7.3,1.4,0.7hz,1h),7.49(dd,j=7.9,1.4hz,1h),7.36(ddd,j=8.1,7.3,1.4hz, 1h),4.96(s,1h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t, j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h).ms(esi):[m+h]449.1618.
[0328]
(3)组分a-7的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 13(90mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-7(1.72g),根据核磁氢谱图,可计算出化合物13的标记率大约为2.38%。
[0329]
实施例八:组分a-8的合成
[0330][0331]
(1)化合物14的合成:按参考文献pauloehrl,t.;delaittre,g.;bruns,m.;meiβler,m.;h.g.;bastmeyer,m.;barner-kowollik,c.angew.chem.int.ed.2012,51,9181.公开的方法进行合成。
[0332]
(2)化合物15的合成:将化合物14(1g,2.6mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物15(0.90g,产率83%)。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.96(s,2h),4.13(t,j=6.1hz, 2h),3.99(s,3h),
3.90-3.80(m,1h),3.63-3.52(m,1h),3.32(dd,j=11.6,5.7hz,2h),2.82 (t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h),2.00-1.34(m,6h).ms(esi): [m+h]412.2027.
[0333]
(3)组分a-8的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 15(82mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物a-8(1.86g),根据核磁氢谱图,可计算出化合物15的标记率大约为3.43%。
[0334]
实施例九:组分a-9的合成
[0335][0336]
(1)化合物16的合成:按参考文献patchornik abraham.;amit b.;woodward r.b.j. am.chem.soc.1970,92,6333.公开的方法进行合成。
[0337]
(2)化合物17的合成:将化合物16(1g,2.5mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物17(0.80g,产率75%)。1h nmr(400mhz,cdcl3):δ=8.02-7.23(m,5h),7.71(s,1h),7.22(s,1h),4.96(s,2h), 4.13(t,j=6.1hz,2h),3.99(s,3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h), 2.44(t,j=7.2hz,2h),2.26-2.17(m,2h).ms(esi):[m+h]432.1713.
[0338]
(3)组分a-9的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 17(86mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-9(1.82g),根据核磁氢谱图,可计算出化合物17的标记率大约为3.24%。
[0339]
实施例十:组分a-10的合成
[0340]
[0341]
(1)化合物18的合成:按参考文献patchornik abraham.;amit b.;woodward r.b.j. am.chem.soc.1970,92,6333.公开的方法进行合成。
[0342]
(2)化合物19的合成:将化合物18(1g,2.7mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物19(0.76g,产率71%)。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.96(s,2h),4.25(q,j=6.5hz, 2h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz, 2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h),1.32(t,j=6.5hz,3h).ms(esi):[m+h] 400.1742.
[0343]
(3)组分a-10的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 19(80mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物a-10(1.88g),根据核磁氢谱图,可计算出化合物19的标记率大约为3.01%。
[0344]
实施例十一:组分a-11的合成
[0345][0346]
(1)化合物20的合成:按参考文献kalbag,s.m.;roeske,r.w.j.am.chem.soc. 1975,97,440.公开的方法进行合成。
[0347]
(2)化合物21的合成:将化合物20(1g,2.3mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物21(0.84g,产率79%)。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.96(s,2h),4.63(q,j=6.9hz, 1h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.67(s,3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t, j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h),1.48(d,j=6.9hz,3h).ms (esi):[m+h]457.1976.
[0348]
(3)组分a-11的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 21(91mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-11(1.76g),根据核磁氢谱图,可计算出化合物21的标记率大约为3.15%。
[0349]
实施例十二:组分a-12的合成
[0350][0351]
(1)化合物22的合成:按参考文献patchornik abraham.;amit b.;woodward r.b.j. am.chem.soc.1970,92,6333.公开的方法进行合成。
[0352]
(2)化合物23的合成:将化合物22(1g,2.7mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物23(0.76g,产率71%)。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.96(s,2h),4.25(q,j=6.5hz, 2h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz, 2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h),1.32(t,j=6.5hz,3h).ms(esi):[m+h] 416.1422.
[0353]
(3)组分a-12的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物23(80mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-12(1.88g),根据核磁氢谱图,可计算出化合物23的标记率大约为3.01%。
[0354]
实施例十三:组分a-13的合成
[0355][0356]
(1)化合物24的合成:按参考文献engels,j.;schlaeger,e.j.j.med.chem.1977,20, 907.公开的方法进行制备化合物24。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s, 1h),4.96(s,2h),4.25(q,j=6.5hz,2h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.32(dd,j= 11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h),1.32(t, j=6.5hz,3h).ms(esi):[m+h]435.1432.
[0357]
(2)组分a-13的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 24(87mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离
子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-13(1.73g),根据核磁氢谱图,可计算出化合物24的标记率大约为3.08%。
[0358]
实施例十四:组分a-14的合成
[0359][0360]
(1)化合物25的合成:按参考文献yunlong yang;jieyuan zhang;zhenzhen liu; qiuning lin;xiaolin liu;chunyan bao;yang wang;linyong zhu.adv.mater.2016,28,2724. 公开的方法进行制备化合物25。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),4.96(s,2h), 4.13(t,j=6.1hz,2h),3.99(s,3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h), 2.44(t,j=7.2hz,2h),2.26-2.17(m,2h).ms(esi):[m+h]362.1124.
[0361]
(2)组分a-14的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 25(72mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-14(1.72g),根据核磁氢谱图,可计算出化合物25的标记率大约为3.09%。
[0362]
实施例十五:组分a-15的合成
[0363][0364]
(1)化合物26的合成:按参考文献yunlong yang;jieyuan zhang;zhenzhen liu; qiuning lin;xiaolin liu;chunyan bao;yang wang;linyong zhu.adv.mater.2016,28,2724. 公开的方法进行制备化合物26。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),4.96(s,2h), 4.13(t,j=6.1hz,2h),3.99(s,3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h), 2.44(t,j=7.2hz,2h),2.35(s,3h),2.26-2.17(m,2h).ms(esi):[m+h]342.1617.
[0365]
(2)组分a-15的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 26(68mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-15(1.88g),根据核磁氢谱图,可
计算出化合物26的标记率大约为3.28%。
[0366]
实施例十六:组分a-16的合成
[0367][0368]
(1)化合物27的合成:按参考文献yunlong yang;jieyuan zhang;zhenzhen liu;qiuning lin;xiaolin liu;chunyan bao;yang wang;linyong zhu.adv.mater.2016,28,2724. 公开的方法进行制备化合物27。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),4.96(s,2h), 4.13(t,j=6.1hz,2h),3.99(s,3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h), 2.76(q,j=6.5hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h),1.22(t,j=6.5hz,3h). ms(esi):[m+h]388.1554.
[0369]
(2)组分a-16的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 27(77mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-16(1.68g),根据核磁氢谱图,可计算出化合物27的标记率大约为2.98%。
[0370]
实施例十七:组分a-17的合成
[0371][0372]
(1)化合物28的合成:按参考文献yunlong yang;jieyuan zhang;zhenzhen liu; qiuning lin;xiaolin liu;chunyan bao;yang wang;linyong zhu.adv.mater.2016,28,2724. 公开的方法进行制备化合物28。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),4.96(s,2h), 4.13(t,j=6.1hz,2h),3.99(s,3h),3.45(q,j=7.26hz,4h),3.32(dd,j=11.6,5.7hz,2h), 2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h),1.20(t,j=7.26hz,6h). ms(esi):[m+h]399.2271.
[0373]
(2)组分a-17的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 28(80mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-17(1.76g),根据核磁氢谱图,可
计算出化合物28的标记率大约为3.02%。
[0374]
实施例十八:组分a-18的合成
[0375][0376]
(1)化合物29的合成:按参考文献yunlong yang;jieyuan zhang;zhenzhen liu; qiuning lin;xiaolin liu;chunyan bao;yang wang;linyong zhu.adv.mater.2016,28,2724. 公开的方法进行制备化合物29。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),4.96(s,2h), 4.13(t,j=6.1hz,2h),3.99(s,3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h), 2.44(t,j=7.2hz,2h),2.26-2.17(m,2h),2.08(s,3h).ms(esi):[m+h]386.1523.
[0377]
(2)组分a-18的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 29(77mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-18(1.82g),根据核磁氢谱图,可计算出化合物29的标记率大约为3.15%。
[0378]
实施例十九:组分a-19的合成
[0379][0380]
(1)化合物30的合成:按参考文献yunlong yang;jieyuan zhang;zhenzhen liu; qiuning lin;xiaolin liu;chunyan bao;yang wang;linyong zhu.adv.mater.2016,28,2724. 公开的方法进行制备化合物30。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),4.96(s,2h), 4.13(t,j=6.1hz,2h),3.99(s,3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h), 2.44(t,j=7.2hz,2h),2.26-2.17(m,2h),2.08(s,3h).ms(esi):[m+h]385.1712.
[0381]
(2)组分a-19的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml
ꢀꢀ
0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 30(77mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-19(1.74g),根据核磁氢谱图,可
计算出化合物30的标记率大约为3.12%。
[0382]
实施例二十:组分a-20的合成
[0383][0384]
(1)化合物31的合成:按参考文献yunlong yang;jieyuan zhang;zhenzhen liu;
[0385]
qiuning lin;xiaolin liu;chunyan bao;yang wang;linyong zhu.adv.mater.2016,28,2724. 公开的方法进行制备化合物31。1h nmr(400mhz,cdcl3):δ=8.02-7.23(m,5h),7.71(s, 1h),4.96(s,2h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t, j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h).ms(esi):[m+h]404.1882.
[0386]
(2)组分a-20的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 31(80mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-20(1.81g),根据核磁氢谱图,可计算出化合物31的标记率大约为3.21%。
[0387]
实施例二十一:组分a-21的合成
[0388][0389]
(1)化合物32的合成:按参考文献emmanuel riguet.;christian g.bochet.org.lett. 2007,26,5453.公开的方法进行合成。
[0390]
(2)化合物33的合成:将化合物32(1g,3.4mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物33(0.85g,产率78%)。1h nmr(400mhz,cdcl3):δ=8.05(d,j=9.54hz,1h),7.24(d,j=2.72hz,1h),6.92(dd, j=9.54,2.72hz,1h),4.85(s,2h),3.56-3.68(m,4h),3.49-3.56(m,2h),3.42-3.49(m, 2h),3.32(t,j=5.9hz,2h),2.82(t,j=5.9hz,2h).ms(esi):[m+h]346.1454.
[0391]
(3)组分a-21的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 33(65mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔
1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-21(1.76g),根据核磁氢谱图,可计算出化合物33的标记率大约为2.84%。
[0392]
实施例二十二:组分a-22的合成
[0393][0394]
(1)化合物34的合成:按参考文献isabelle aujard.;chouaha benbrahim.;ludovicjullien.chem.eur.j.2006,12,6865.公开的方法进行合成。
[0395]
(2)化合物35的合成:将化合物34(1g,3.2mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物35(0.96g,产率88%)。1h nmr(400mhz,cdcl3):δ=8.05(d,j=9.54hz,1h),7.28(d,j=8.00hz,2h),7.24(d,j =2.72hz,1h),6.92(dd,j=9.54,2.72hz,1h),6.78(d,8.00hz,2h),4.96(s,2h),4.83(s, 2h),3.32(t,j=5.9hz,2h),2.82(t,j=5.9hz,2h).ms(esi):[m+h]346.1454.
[0396]
(3)组分a-22的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 35(69mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-22(1.83g),根据核磁氢谱图,可计算出化合物35的标记率大约为3.12%。
[0397]
实施例二十三:组分a-23的合成
[0398][0399]
(1)化合物36的合成:按参考文献filiz bley.;klaus schaper.;helmutgorner.photochem.photobiol.2008,84,162.公开的方法进行合成。
[0400]
(2)化合物37的合成:将化合物36(1g,3.5mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物37(0.83g,产率76%)。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),5.92(s,2h),4.96(s,2h),4.83(s,2h),3.32(t,j =5.9hz,2h),2.82(t,j=5.9hz,2h).ms(esi):[m+h]314.0932.
[0401]
(3)组分a-23的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml
ꢀꢀ
0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 37(63mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-23(1.84g),根据核磁氢谱图,可计算出化合物37的标记率大约为3.17%。
[0402]
实施例二十四:组分a-24的合成
[0403][0404]
(1)化合物38的合成:按参考文献filiz bley.;klaus schaper.;helmutgorner.photochem.photobiol.2008,84,162.公开的方法进行合成。
[0405]
(2)化合物39的合成:将化合物38(1g,3.0mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物39(0.77g,产率71%)。1h nmr(400mhz,cdcl3):δ=4.96(s,2h),4.83(s,2h),3.32(t,j=5.9hz,2h),3.27-3.21 (m,4h),2.84(t,j=6.5hz,2h),2.82(t,j=5.9hz,2h),2.75(t,j=6.3hz,2h),2.00-1.91(m, 4h).ms(esi):[m+h]365.1828.
[0406]
(3)组分a-24的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 39(73mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-24(1.87g),根据核磁氢谱图,可计算出化合物39的标记率大约为3.06%。
[0407]
实施例二十五:组分a-25的合成
[0408][0409]
(1)化合物40的合成:按参考文献emmanuel riguet.;christian g.bochet.org.lett. 2007,26,5453.公开的方法进行合成。
[0410]
(2)化合物41的合成:将化合物40(1g,3.6mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物41(0.93g,产率85%)。1h nmr(400mhz,
cdcl3):δ=7.71(s,1h),7.22(s,1h),4.96(s,2h),4.24(s,2h),3.32(t,j =5.9hz,2h),3.27-3.21(m,2h),2.82(t,j=5.9hz,2h),2.75(t,j=6.3hz,2h),2.00-1.91 (m,2h).ms(esi):[m+h]309.1522.
[0411]
(3)组分a-25的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 41(62mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-25(1.82g),根据核磁氢谱图,可计算出化合物41的标记率大约为3.12%。
[0412]
实施例二十六:组分a-26的合成
[0413][0414]
(1)化合物42的合成:按参考文献singh,a.k.;khade,p.k.tetrahedron.2005,61, 10007.公开的方法进行合成。
[0415]
(2)化合物43的合成:将化合物42(1g,3.4mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物43(0.90g,产率82%)。1h nmr(400mhz,cdcl3):δ=8.31-7.12(m,5h),4.96(s,2h),4.83(s,2h),3.32(t,j=5.9 hz,2h),2.82(t,j=5.9hz,2h).ms(esi):[m+h]320.1254.
[0416]
(3)组分a-26的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 43(64mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-26(1.87g),根据核磁氢谱图,可计算出化合物43的标记率大约为3.21%。
[0417]
实施例二十七:组分a-27的合成
[0418][0419]
(1)化合物44的合成:按参考文献felix friedrich.;mike heilemann.;alexander heckel. chem.commun.2015,51,15382.公开的方法进行合成。
[0420]
(2)化合物45的合成:将化合物44(1g,3.0mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物45(0.80g,产率74%)。1h nmr(400mhz,cdcl3):δ=8.31-7.12(m,5h),4.96(s,2h),4.83(s,2h),3.32(t,j=5.9 hz,2h),2.82(t,j=5.9hz,2h).ms(esi):[m+h]360.1254.
[0421]
(3)组分a-27的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 45(72mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-27(1.78g),根据核磁氢谱图,可计算出化合物45的标记率大约为2.12%。
[0422]
实施例二十八:组分a-28的合成
[0423][0424]
(1)化合物46的合成:按参考文献grazyna groszek.;agnieszka nowak-krol.;barbarafilipek.eur.j.med.chem.2009,44,5103.公开的方法进行合成。
[0425]
(2)化合物47的合成:将化合物46(1g,3.3mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物47(0.97g,产率89%)。1h nmr(400mhz,cdcl3):δ=8.04(s,1h),7.42(s,1h),4.96(s,2h),4.13(t,j=6.1hz, 2h),3.99(s,3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz, 2h),2.26-2.17(m,2h).ms(esi):[m+h]328.1507.
[0426]
(3)组分a-28的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 47(65mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-28(1.85g),根据核磁氢谱图,可计算出化合物47的标记率大约为3.43%。
[0427]
实施例二十九:组分a-29的合成
[0428][0429]
(1)化合物48的合成:按参考文献thomas f.greene.;shu wang.;mary j.meegan.j. med.chem.2016,59,90.公开的方法进行合成。
[0430]
(2)化合物49的合成:将化合物48(1g,3.3mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物49(0.95g,产率87%)。1h nmr(400mhz,cdcl3):δ=7.95(s,1h),7.12(s,1h),4.96(s,2h),4.13(t,j=6.1hz, 2h),3.99(s,3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz, 2h),2.26-2.17(m,2h).ms(esi):[m+h]328.1507.
[0431]
(3)组分a-29的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 49(65mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-29(1.86g),根据核磁氢谱图,可计算出化合物49的标记率大约为3.52%。
[0432]
实施例三十:组分a-30的合成
[0433][0434]
(1)化合物50的合成:按参考文献yu-shan.;mohane selvaraj coumar.;hsing-panghsieh.j.med.chem.2009,52,4941.公开的方法进行合成。
[0435]
(2)化合物51的合成:将化合物50(1g,3.3mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物51(0.89g,产率81%)。1h nmr(400mhz,cdcl3):δ=7.64(s,1h),7.02(s,1h),4.96(s,2h),4.13(t,j=6.1hz, 2h),3.99(s,3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz, 2h),2.26-2.17(m,2h).ms(esi):[m+h]328.1507.
[0436]
(3)组分a-30的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 51(65mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔
1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-30(1.82g),根据核磁氢谱图,可计算出化合物51的标记率大约为3.39%。
[0437]
实施例三十一:组分a-31的合成
[0438][0439]
(1)化合物52的合成:按参考文献sarit s.agasti.;apiwat chompoosor.;vincent m. rotello.j.am.chem.soc.2009,131,5728.公开的方法进行合成。
[0440]
(2)化合物53的合成:将化合物52(1g,2.9mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物53(0.91g,产率84%)。1h nmr(400mhz,cdcl3):δ=7.91(s,1h),4.96(s,2h),4.13(t,j=6.1hz,2h),3.99(s, 3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26
-ꢀ
2.17(m,2h).ms(esi):[m+h]373.1373.
[0441]
(3)组分a-31的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 53(75mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-31(1.87g),根据核磁氢谱图,可计算出化合物53的标记率大约为3.45%。
[0442]
实施例三十二:组分a-32的合成
[0443][0444]
(1)化合物54的合成:按参考文献chandramouleeswaran subramani.;xi yu.;vincentm.rotello.j.mater.chem.2011,21,14156.公开的方法进行合成。
[0445]
(2)化合物55的合成:将化合物54(1g,2.7mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物55(0.84g,产率78%)。1h nmr(400mhz,cdcl3):δ=4.96(s,2h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.32(dd,j =11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h).ms (esi):[m+h]403.1432.
[0446]
(3)组分a-32的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物55(80mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-32(1.81g),根据核磁氢谱图,可计算出化合物55的标记率大约为3.09%。
[0447]
实施例三十三:组分a-33的合成
[0448][0449]
组分a-33的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml 0.01mol/l2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取nb混合物(化合物1/化合物55,60mg,重量比1:1)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6-二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3mlmes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物a-33(1.87g),根据核磁氢谱图,可计算出nb混合物(化合物1/化合物55) 的标记率大约为3.52%。
[0450]
实施例三十四:组分a-34的合成
[0451][0452]
(1)化合物56的合成:按参考文献pauloehrl,t.;delaittre,g.;bruns,m.;meiβler,m.;h.g.;bastmeyer,m.;barner-kowollik,c.angew.chem.int.ed.2012,51,9181.公开的方法进行合成。
[0453]
(2)化合物57的合成:将化合物56(1g,3.3mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物57(0.93g,产率85%)。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.96(s,2h),3.99(s,3h),3.32(t,j =5.7hz,2h),2.82(t,j=5.9hz,2h),2.55(t,j=6.1hz,2h),2.44(t,j=7.2hz,2h),2.26
-ꢀ
2.17(m,2h).ms(esi):[m+h]326.1721.
[0454]
(3)组分a-34的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 57(65mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三
嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-34(1.82g),根据核磁氢谱图,可计算出化合物57的标记率大约为3.21%。
[0455]
实施例三十五:组分a-35的合成
[0456][0457]
(1)化合物58的合成:按参考文献pauloehrl,t.;delaittre,g.;bruns,m.;meiβler,m.;h.g.;bastmeyer,m.;barner-kowollik,c.angew.chem.int.ed.2012,51,9181.公开的方法进行合成。
[0458]
(2)化合物59的合成:将化合物58(1g,3.3mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶解-重沉淀后,过滤、真空干燥即可得到化合物59(0.82g,产率75%)。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.96(s,2h),4.03(t,j=6.1hz, 2h),3.99(s,3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz, 2h),2.26-2.17(m,2h).ms(esi):[m+h]360.1213.
[0459]
(3)组分a-35的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 59(65mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-35(1.87g),根据核磁氢谱图,可计算出化合物59的标记率大约为2.76%。
[0460]
实施例三十六:组分a-36的合成
[0461][0462]
(1)化合物60的合成:按参考文献pauloehrl,t.;delaittre,g.;bruns,m.;meiβler,m.;h.g.;bastmeyer,m.;barner-kowollik,c.angew.chem.int.ed.2012,51,9181.公开的方法进行合成。
[0463]
(2)化合物61的合成:将化合物60(1g,3.3mmol)和乙二胺(1.1ml)溶于甲醇(50ml)中,回流过夜反应后,减压旋蒸,将粗产物溶于甲醇中,在乙酸乙酯中重沉淀。经过多次溶
解-重沉淀后,过滤、真空干燥即可得到化合物61(0.80g,产率73%)。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.96(s,2h),3.99(s,3h),3.45(t,j =6.1hz,2h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h), 2.26-2.17(m,2h).ms(esi):[m+h]327.1625.
[0464]
(3)组分a-36的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 61(65mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-36(1.76g),根据核磁氢谱图,可计算出化合物61的标记率大约为3.21%。
[0465]
实施例三十七:组分a-37的合成
[0466][0467]
组分a-37的合成:将羧甲基纤维素carboxymethyl cellulose(2g,90kda)溶于100 ml 0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物1(65mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏羧甲基纤维素衍生物a-37(1.89g),根据核磁氢谱图,可计算出化合物1的标记率大约为2.25%。
[0468]
实施例三十八:组分a-38的合成
[0469][0470]
组分a-38的合成:将海藻酸alginic acid(2g)溶于100ml 0.01mol/l 2-(n-吗啉) 乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物1(65mg,0.2mmol) 溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6-二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h) 加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏海藻酸衍生物a-38(1.82g),根据核磁氢谱图,可计算出化合物1的标记率大约为3.17%。
[0471]
实施例三十九:组分a-39的合成
[0472][0473]
组分a-39的合成:将硫酸软骨素chondroitin sulfate(2g)溶于100ml 0.01mol/l 2
-ꢀ
(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物1(65mg, 0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6-二甲氧基三嗪
ꢀ-
2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次 (每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000) 中,用去离子水透析2-3d,冷冻干燥即可得到光敏硫酸软骨素衍生物a-39(1.73g),根据核磁氢谱图,可计算出化合物1的标记率大约为2.98%。
[0474]
实施例四十:组分a-40的合成
[0475][0476]
组分a-40的合成:将聚谷氨酸pga(1g)溶于50ml蒸馏水中至完全溶解,加入羟基苯并三唑(hobt,0.3g,2.3mmol),然后将溶于甲醇中的化合物1(0.5g,1.6mmol) 和1-乙基-(3-二甲基胺基丙基)碳二亚胺盐酸盐(edc-hcl,0.5g,2.6mmol)加入到上述溶液中室温反应48h后,先用含氯化钠的稀盐酸溶液(ph=3.5)透析1d,再用纯水透析1d后,冷冻干燥即可得到光敏聚谷氨酸衍生物a-40(0.92g),根据其核磁氢谱图,可以计算出化合物1的修饰度大约为21.3%。
[0477]
实施例四十一:组分a-41的合成
[0478][0479]
组分a-41的合成:将四臂聚乙二醇羧酸衍生物4-peg-cooh(0.5g,10kda)溶于 20ml无水二甲基亚砜dmso中至完全溶解,取化合物1(130mg,0.4mmol)溶于5ml 无水二甲基亚砜dmso后加入上述反应液,加入0.2ml三乙胺tea,再加入六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷pybop(210mg,0.4mmol),于室温下反应24h,然后在乙醚中重沉淀,将粗产物溶于水后倒入透析袋(mwco 3500)中,用去离子水透析2-3d,冷冻干燥即可得到光敏聚乙二醇衍生物a-41(0.45g),根据核磁氢谱图,可计算出化合物1的标记率大约为98%。
[0480]
实施例四十二:组分a-42的合成
[0481][0482]
(1)化合物62的合成:按参考文献pauloehrl,t.;delaittre,g.;bruns,m.;meiβler,m.;h.g.;bastmeyer,m.;barner-kowollik,c.angew.chem.int.ed.2012,51,9181.公开的方法进行合成。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.96(s,2h), 4.13(t,j=6.1hz,2h),3.99(s,3h),3.90-3.80(m,1h),3.79(t,j=6.1hz,2h),3.70(t,j= 7.2hz,2h),3.63-3.52(m,1h),3.56(t,j=7.2hz,2h),2.00-1.34(m,6h).ms(esi):[m+h] 372.1627.
[0483]
(2)组分a-42的合成:将透明质酸hyaluronic acid(1g,340kda)溶于50ml水中,将化合物62(0.2g,0.48mmol)、edc-hcl(0.76g,3.96mmol)和dpts(0.12g, 0.48mmol)依次加入到上述溶液中,室温下搅拌反应48h。反应结束后,将反应液倒入冷的乙醇中多次重沉淀纯化,收集到的沉淀干燥后将其溶于无水dmso中,加入对甲苯磺酸将二氢吡喃保护基团脱掉即可得到光敏透明质酸衍生物a-42(0.86g)。根据其核磁氢谱图,可计算出化合物62的修饰度大约为10%。
[0484]
实施例四十三:组分a-43的合成
[0485][0486]
(1)化合物63的合成:按参考文献pauloehrl,t.;delaittre,g.;bruns,m.;meiβler,m.;h.g.;bastmeyer,m.;barner-kowollik,c.angew.chem.int.ed.2012,51,9181.公开的方法进行合成。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.96(s,2h), 4.13(t,j=6.1hz,2h),3.99(s,3h),3.90-3.80(m,1h),3.63-3.52(m,1h),2.44(t,j=7.2 hz,2h),2.26-2.17(m,2h),2.00-1.34(m,6h).ms(esi):[m+h]370.1512.
[0487]
(2)组分a-43的合成:将1g壳聚糖加入到75ml异丙醇中形成壳聚糖的悬浮液,然后将化合物63(0.2g,0.54mmol)、edc-hcl(0.76g,3.96mmol)和nhs(0.46g, 4.0mmol)依次加入到上述溶液中,室温下搅拌反应48h。反应结束后,将混合物溶液过滤,滤液用甲醇/水混合溶剂透析三次、甲醇透析两次后,冷冻干燥即可得到化合物63标记的壳聚糖(0.9g)。将化合物63标记的壳聚糖溶于dmso中,加入对甲苯磺酸脱除二氢吡喃保护即可得到光敏壳聚糖衍生物a-43,根据其核磁氢谱图,可计算出化合物63的修饰度大约为12.5%。
[0488]
实施例四十四:组分a-44的合成
[0489][0490]
组分a-44的合成:将聚赖氨酸pll(1g)溶于50ml水中,将化合物63(0.2g, 0.54mmol)、edc-hcl(0.76g,3.96mmol)和nhs(0.46g,4.0mmol)依次加入到上述溶液中,室温下搅拌反应48h。反应结束后,将反应液倒入冷的乙醇中多次重沉淀纯化,收集到的沉淀干燥后将其溶于无水dmso中,加入对甲苯磺酸将二氢吡喃保护基团脱掉即可得到光敏聚赖氨酸衍生物a-44(0.84g)。根据其核磁氢谱图,可计算出化合物63的修饰度大约为15.6%。
[0491]
实施例四十五:组分a-45的合成
[0492][0493]
组分a-45的合成:将明胶gelatin(1g)溶于50ml蒸馏水中至完全溶解,将化合物 63(0.2g,0.54mmol)、edc-hcl(0.76g,3.96mmol)和nhs(0.46g,4.0mmol)依次加入到上述溶液中,室温下搅拌反应48h。反应结束后,将反应液倒入冷的乙醇中多次重沉淀纯化,收集到的沉淀干燥后将其溶于无水dmso中,加入对甲苯磺酸将二氢吡喃保护基团脱掉即可得到光敏明胶衍生物a-45(0.83g),根据其核磁氢谱图,可以计算出化合物63的修饰度大约为11.2%。
[0494]
实施例四十六:组分a-46的合成
[0495][0496]
组分a-46的合成:将葡聚糖dextran(1g)溶于50ml水中,将化合物63(0.23g, 0.54mmol)、edc-hcl(0.76g,3.96mmol)和dpts(0.12g,0.48mmol)依次加入到上述溶液中,室温下搅拌反应48h。反应结束后,将反应液倒入冷的乙醇中多次重沉淀纯化,收集到的沉淀干燥后将其溶于无水dmso中,加入对甲苯磺酸将二氢吡喃保护基团脱掉即可得到光敏葡聚糖衍生物a-46(0.92g)。根据其核磁氢谱图,可计算出化合物63 的修饰度大约为18.2%。
[0497]
实施例四十七:组分a-47的合成
[0498][0499]
(1)化合物64的合成:按参考文献pauloehrl,t.;delaittre,g.;bruns,m.;meiβler,m.;h.g.;bastmeyer,m.;barner-kowollik,c.angew.chem.int.ed.2012,51,9181.公开的方法进行合成。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.96(s,2h), 4.13(t,j=6.1hz,2h),3.99(s,3h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h).ms(esi): [m+h]286.0943.
[0500]
(2)组分a-47的合成:将巯基修饰的肝素hep-sh(1g)溶于50ml蒸馏水中至完全溶解,加入羟基苯并三唑(hobt,0.3g,2.3mmol),然后将溶于甲醇中的化合物64(0.5 g,1.6mmol)和1-乙基-(3-二甲基胺基丙基)碳二亚胺盐酸盐(edc-hcl,0.5g,2.6mmol) 加入到上述溶液中室温反应48h后,先用含氯化钠的稀盐酸溶液(ph=3.5)透析1d,再用纯水透析1d后,冷冻干燥即可得到光敏肝素衍生物a-47(0.86g),根据其核磁氢谱图,可以计算出化合物64的修饰度大约为10.2%。
[0501]
实施例四十八:组分a-48的合成
[0502][0503]
(1)化合物65的合成:按参考文献pauloehrl,t.;delaittre,g.;bruns,m.;meiβler,m.;h.g.;bastmeyer,m.;barner-kowollik,c.angew.chem.int.ed.2012,51,9181.公开的方法进行合成。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.96(s,2h), 4.13(t,j=6.1hz,2h),3.99(s,3h),3.90-3.80(m,1h),3.63-3.52(m,1h),3.04(t,j=7.2 hz,2h),2.00-1.34(m,6h).ms(esi):[m+h]391.0518.
[0504]
(2)组分a-48的合成:将1g壳聚糖加入到75ml异丙醇中形成壳聚糖的悬浮液, 25ml的naoh溶液(10mol/l)分五次慢慢加入到上述壳聚糖的悬浮液中并继续搅拌半小时左右。然后将化合物65(0.2g)加入到上述溶液中并在60℃条件下反应3h。反应结束后,将混合物溶液过滤,滤液用甲醇/水混合溶剂透析三次、甲醇透析两次后,冷冻干燥即可得到化合物65标记的壳聚糖(0.92g)。将化合物65标记的壳聚糖溶于dmso中,加入对甲苯磺酸脱除二氢吡喃保护即可得到光敏壳聚糖衍生物a-48(0.84g),根据其核磁氢谱图,可计算出化合物65的修饰度大约为12.4%。
[0505]
实施例四十九:组分a-49的合成
[0506][0507]
组分a-49的合成:将peg-4oh(1g,0.05mmol)溶于无水乙腈中,加入k2co3(55.3 mg,0.4mmol)搅拌30min后,加入化合物65(0.17g,0.4mmol)于室温下继续反应 24h。反应结束后,将大部分溶剂除掉,在乙醚中重沉淀,并多次洗涤,然后将化合物65 标记的聚乙二醇溶于dmso中,加入对甲苯磺酸脱除二氢吡喃保护即可得到光敏聚乙二醇衍生物a-49(0.93g),根据核磁氢谱图,可计算出化合物65的修饰度大约为95%。
[0508]
实施例五十:组分a-50的合成
[0509][0510]
(1)化合物66的合成:将化合物65(0.5g,1.29mmol)和乙二醇(0.24g,3.87mmol) 溶于无水乙腈中,加入k2co3(0.5g,3.87mmol)做碱,回流过夜反应。待反应结束后,减压旋蒸掉溶剂,过柱纯化,即可得到化合物66(0.34g,72%)。1h nmr(400mhz,cdcl3): δ=7.71(s,1h),7.22(s,1h),4.96(s,2h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.90-3.80(m, 1h),3.79(t,j=6.1hz,2h),3.70(t,j=7.2hz,2h),3.63-3.52(m,1h),3.56(t,j=7.2hz, 2h),2.00-1.34(m,6h).ms(esi):[m+h]372.1627.
[0511]
(2)化合物67的合成:将化合物66(0.64g,1.72mmol)和三乙胺(0.34g,3.44mmol) 溶于干燥的二氯甲烷中,冰浴条件下,甲基丙烯酰氯(0.27g,2.58mmol)慢慢逐滴加入到上述溶液中,滴加完后室温条件下过夜反应。反应结束后,减压旋蒸掉溶剂,过柱纯化即可得到化合物67(0.49g,65%)。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h), 6.25(s,1h),5.68(s,1h),4.96(s,2h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.90-3.80(m, 1h),3.79(t,j=6.1hz,2h),3.70(t,j=7.2hz,2h),3.63-3.52(m,1h),3.56(t,j=7.2hz, 2h),2.00-1.34(m,6h),1.87(s,3h).ms(esi):[m+h]440.1942.
[0512]
(3)组分a-50的合成:称取化合物67(0.28g,0.63mmol)、共单体peg-ma(0.882 g,2.52mmol)和引发剂偶氮二异丁腈(11mg)加入到史莱克管中,并加入无水thf溶解,经过多次冷冻-抽真空循环操作处理后,将该反应体系于75℃条件下反应24h。反应结束后,将反应液倒入冷的乙醚中多次重沉淀纯化,收集到的沉淀干燥后将其溶于无水 dmso中,加入对甲苯磺酸将二氢吡喃保护基团脱掉即可得到光敏共聚物衍生物a-50 (0.84g)。根据核磁氢谱图,可计算出化合物67在共聚物中的含量大约为15.5%。根据 gpc测得合成高分子的分子量在25kda左右,根据投料比计算可得n为12,x为10,y 为40。
[0513]
实施例五十一:组分a-51的合成
[0514][0515]
(1)化合物68的合成:按参考文献kunihiko morihiro.;tetsuya kodama.;shohei mori.; satoshi obika.org.biomol.chem.2014,12,2468.公开的方法进行合成。1h nmr(400mhz, cdcl3):δ=7.71(s,1h),7.22(s,1h),4.03(s,2h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.32 (dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h). ms(esi):[m+h]344.1207.
[0516]
(2)组分a-51的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 68(69mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-51(1.85g),根据核磁氢谱图,可计算出化合物68的标记率大约为3.34%。
[0517]
实施例五十二:组分a-52的合成
[0518][0519]
(1)化合物69的合成:按参考文献yunlong yang;jieyuan zhang;zhenzhen liu; qiuning lin;xiaolin liu;chunyan bao;yang wang;linyong zhu.adv.mater.2016,28,2724. 公开的方法进行合成。
[0520]
(2)化合物70的合成:将化合物69(1g,3.0mmol)溶于50ml四氢呋喃中,分别加入四溴化碳cbr4(2g,6.0mmol),和三苯基膦pph3(1.6g,6.0mmol),氩气保护下,室温搅拌反应2h,反应后加入5ml水淬灭反应,旋干溶剂,用乙酸乙酯萃取,柱层析分离(pe:dcm=4:1),即可得到化合物70(1.0g,产率84%)。1h nmr(400mhz, cdcl3):δ=7.71(s,1h),7.22(s,1h),4.56(s,2h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.32 (dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h). ms(esi):[m+h]390.0623.
[0521]
(3)化合物71的合成:将化合物70(0.5g,1.3mmol)溶于50ml丙酮中,分别加入l-半胱氨酸甲酯盐酸盐(0.45g,2.6mmol),和氢氧化钠(0.2g,5.2mmol),氩气保护下,室温搅拌反应2h,反应后加入4m hcl调节ph=7,旋干溶剂,用乙酸乙酯萃取,柱层析分离(pe:dcm=4:1),即可得化合物71(0.7g,产率88%)。1h nmr(400 mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.76(s,2h),4.42(m,1h),4.13(t,j=6.1hz,2h), 3.99(s,3h),3.95(s,3h),3.43(d,j
=5.6,2h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9 hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h),1.42(s,9h).ms(esi):[m+h] 545.2219.
[0522]
(4)组分a-52的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 71(109mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-52(1.92g),根据核磁氢谱图,可计算出化合物71的标记率大约为3.32%。
[0523]
实施例五十三:组分a-53的合成
[0524][0525]
(1)化合物72的合成:按参考文献james f.cameron.;jean m.j.frechet.j.am.chem. soc.1991,113,4303.公开的方法进行合成。
[0526]
(2)化合物73的合成:按实施例52的方法,以化合物72为原料制得化合物73(产率73%)。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.66(m,1h),4.13(t,j= 6.1hz,2h),3.99(s,3h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j= 7.2hz,2h),2.26-2.17(m,2h),1.33(d,j=6.9hz,3h).ms(esi):[m+h]404.0863.
[0527]
(3)化合物74的合成:按实施例52的方法,以化合物73为原料制得化合物74(产率70%)。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.86(m,1h),4.42(m, 1h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.95(s,3h),3.43(d,j=5.6,2h),3.32(dd,j=11.6, 5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h),1.42(s,9h), 1.33(d,j=6.9hz,3h).ms(esi):[m+h]559.2402.
[0528]
(4)组分a-53的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 74(112mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-53(1.75g),根据核磁氢谱图,可计算出化合物74的标记率大约为2.34%。
[0529]
实施例五十四:组分a-54的合成
[0530][0531]
(1)化合物75的合成:按参考文献jack e.baldwin.;adrian w.mcconnaughie.;sungbo shin.tetrahedron.1990,46,6879.公开的方法进行合成。
[0532]
(2)化合物76的合成:按实施例52的方法,以化合物75为原料制得化合物76(产率64%)。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.75(ddd,j=8.2,1.4,0.4hz,1h), 7.22(s,1h),7.57(tdd,j=7.3,1.4,0.7hz,1h),7.49(dd,j=7.9,1.4hz,1h),7.36(ddd,j= 8.1,7.3,1.4hz,1h),4.66(s,1h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.32(dd,j=11.6,5.7 hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h).ms(esi):[m+h] 511.0881.
[0533]
(3)化合物77的合成:按实施例52的方法,以化合物76为原料制得化合物77(产率58%)。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.75(ddd,j=8.2,1.4,0.4hz,1h), 7.22(s,1h),7.57(tdd,j=7.3,1.4,0.7hz,1h),7.49(dd,j=7.9,1.4hz,1h),7.36(ddd,j= 8.1,7.3,1.4hz,1h),4.86(s,1h),4.42(m,1h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.95(s, 3h),3.43(d,j=5.6,2h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j= 7.2hz,2h),2.26-2.17(m,2h),1.42(s,9h).ms(esi):[m+h]666.2423.
[0534]
(4)组分a-54的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 77(133mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物a-54(1.8g),根据核磁氢谱图,可计算出化合物77的标记率大约为3.35%。
[0535]
实施例五十五:组分a-55的合成
[0536][0537]
(1)化合物78的合成:按参考文献pauloehrl,t.;delaittre,g.;bruns,m.;meiβler,m.;h.g.;bastmeyer,m.;barner-kowollik,c.angew.chem.int.ed.2012,51,9181.公开的方法进行合成。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.76(s,2h), 4.13(t,j=6.1hz,2h),3.99(s,3h),3.90-3.80(m,1h),3.63-3.52(m,1h),3.32(dd,j
=11.6, 5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h),2.00-1.34 (m,6h).ms(esi):[m+h]428.1831.
[0538]
(2)组分a-55的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 78(85mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-55(1.89g),根据核磁氢谱图,可计算出化合物78的标记率大约为3.42%。
[0539]
实施例五十六:组分a-56的合成
[0540][0541]
(1)化合物79的合成:按参考文献patchornik abraham.;amit b.;woodward r.b.j. am.chem.soc.1970,92,6333.公开的方法进行合成。1h nmr(400mhz,cdcl3):δ=8.02
ꢀ-
7.23(m,5h),7.71(s,1h),7.22(s,1h),4.76(s,2h),4.13(t,j=6.1hz,2h),3.99(s,3h), 3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m, 2h).ms(esi):[m+h]448.1561.
[0542]
(2)组分a-56的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 79(89mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-56(1.87g),根据核磁氢谱图,可计算出化合物79的标记率大约为3.21%。
[0543]
实施例五十七:组分a-57的合成
[0544][0545]
(1)化合物80的合成:按参考文献patchornik abraham.;amit b.;woodward r.b.j. am.chem.soc.1970,92,6333.公开的方法进行合成。1h nmr(400mhz,cdcl3):δ=7.71 (s,1h),7.22(s,1h),4.76(s,2h),4.25(q,j=6.5hz,2h),4.13(t,j=6.1hz,2h),
3.99(s,3h), 3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m, 2h),1.32(t,j=6.5hz,3h).ms(esi):[m+h]416.1432.
[0546]
(2)组分a-57的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 80(83mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-57(1.74g),根据核磁氢谱图,可计算出化合物80的标记率大约为2.34%。
[0547]
实施例五十八:组分a-58的合成
[0548][0549]
(1)化合物81的合成:按参考文献kalbag,s.m.;roeske,r.w.j.am.chem.soc. 1975,97,440.公开的方法进行合成。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s, 1h),4.76(s,2h),4.63(q,j=6.9hz,1h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.67(s,3h), 3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m, 2h),1.48(d,j=6.9hz,3h).ms(esi):[m+h]473.1734.
[0550]
(2)组分a-58的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物81(94mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-58(1.72g),根据核磁氢谱图,可计算出化合物81的标记率大约为2.56%。
[0551]
实施例五十九:组分a-59的合成
[0552][0553]
(1)化合物82的合成:按参考文献patchornik abraham.;amit b.;woodward r.b.j. am.chem.soc.1970,92,6333.公开的方法进行合成。1h nmr(400mhz,cdcl3):δ=7.71 (s,1h),7.22(s,1h),4.76(s,2h),4.25(q,j=6.5hz,2h),4.13(t,j=6.1hz,2h),
3.99(s,3h), 3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m, 2h),1.32(t,j=6.5hz,3h).ms(esi):[m+h]432.1224.
[0554]
(2)组分a-59的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 82(83mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-59(1.74g),根据核磁氢谱图,可计算出化合物82的标记率大约为2.34%。
[0555]
实施例六十:组分a-60的合成
[0556][0557]
(1)化合物83的合成:按参考文献engels,j.;schlaeger,e.j.j.med.chem.1977,20, 907.公开的方法进行合成。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.76(s, 2h),4.25(q,j=6.5hz,2h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.32(dd,j=11.6,5.7hz, 2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h),1.32(t,j=6.5hz, 3h).ms(esi):[m+h]451.1126.
[0558]
(2)组分a-60的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 83(90mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-60(1.72g),根据核磁氢谱图,可计算出化合物83的标记率大约为2.36%。
[0559]
实施例六十一:组分a-61的合成
[0560][0561]
(1)化合物84的合成:按实施例52的方法,采用常规化学手段制备化合物84。1h
ꢀꢀ
nmr(400mhz,cdcl3):δ=7.71(s,1h),4.76(s,2h),4.42(m,1h),4.13(t,j=6.1hz,2h), 3.99(s,3h),3.95(s,3h),3.43(d,j=5.6,2h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=
5.9 hz,2h),2.44(t,j=7.2hz,2h),2.35(s,3h),2.26-2.17(m,2h),1.42(s,9h).ms(esi): [m+h]559.2452.
[0562]
(2)组分a-61的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 84(111mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-61(1.73g),根据核磁氢谱图,可计算出化合物84的标记率大约为3.02%。
[0563]
实施例六十二:组分a-62的合成
[0564][0565]
(1)化合物85的合成:按参考文献grazyna groszek.;agnieszka nowak-krol.;barbara filipek.eur.j.med.chem.2009,44,5103.公开的方法进行合成。1h nmr(400mhz, cdcl3):δ=8.04(s,1h),7.42(s,1h),4.76(s,2h),4.42(m,1h),4.13(t,j=6.1hz,2h),3.99 (s,3h),3.95(s,3h),3.43(d,j=5.6,2h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz, 2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h),1.42(s,9h).ms(esi):[m+h]545.2234.
[0566]
(2)组分a-62的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 85(109mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-62(1.92g),根据核磁氢谱图,可计算出化合物85的标记率大约为3.14%。
[0567]
实施例六十三:组分a-63的合成
[0568][0569]
(1)化合物86的合成:按参考文献thomas f.greene.;shu wang.;mary j.meegan.j. med.chem.2016,59,90.公开的方法进行合成。1h nmr(400mhz,cdcl3):δ=7.95(s,1h), 7.12(s,1h),4.76(s,2h),4.42(m,1h),4.13(t,j=6.1hz,2h),3.99(s,3h),
3.95(s,3h),3.43 (d,j=5.6,2h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h), 2.26-2.17(m,2h),1.42(s,9h).ms(esi):[m+h]545.2262.
[0570]
(2)组分a-63的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 86(109mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-63(1.88g),根据核磁氢谱图,可计算出化合物86的标记率大约为3.45%。
[0571]
实施例六十四:组分a-64的合成
[0572][0573]
(1)化合物87的合成:按参考文献yu-shan.;mohane selvaraj coumar.;hsing-pang hsieh.j.med.chem.2009,52,4941.公开的方法进行合成。1h nmr(400mhz,cdcl3):δ= 7.64(s,1h),7.02(s,1h),4.76(s,2h),4.42(m,1h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.95 (s,3h),3.43(d,j=5.6,2h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j =7.2hz,2h),2.26-2.17(m,2h),1.42(s,9h).ms(esi):[m+h]545.2231.
[0574]
(2)组分a-64的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 87(109mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-64(1.85g),根据核磁氢谱图,可计算出化合物87的标记率大约为3.32%。
[0575]
实施例六十五:组分a-65的合成
[0576][0577]
组分a-65的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml 0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取nb混合物(化合物68/化合物71,60mg,质量比1:1)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6-二
甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3mlmes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物a-65(1.89g),根据核磁氢谱图,可计算出nb混合物(化合物68/化合物71) 的标记率大约为3.41%。
[0578]
实施例六十六:组分a-66的合成
[0579][0580]
组分a-66的合成:将羧甲基纤维素carboxymethyl cellulose(2g,340kda)溶于100 ml 0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物71(109mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏羧甲基纤维素衍生物a-66(1.74g),根据核磁氢谱图,可计算出化合物71的标记率大约为2.34%。
[0581]
实施例六十七:组分a-67的合成
[0582][0583]
(1)化合物88的合成:按实施例52的方法,采用常规化学手段制备化合物88。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.76(s,2h),4.42(m,1h),4.13(t,j= 6.1hz,2h),3.99(s,3h),3.95(s,3h),3.43(d,j=5.6,2h),2.44(t,j=7.2hz,2h),2.26-2.17 (m,2h),1.42(s,9h).ms(esi):[m+h]503.1732.
[0584]
(2)组分a-67的合成:将1g壳聚糖加入到75ml异丙醇中形成壳聚糖的悬浮液,然后将化合物88(0.2g,0.40mmol)、edc-hcl(0.76g,3.96mmol)和nhs(0.46g,4.0mmol)依次加入到上述溶液中,室温下搅拌反应48h。反应结束后,先用含氯化钠的稀盐酸溶液(ph=3.5)透析1d,再用纯水透析1d后,冷冻干燥即可得到光敏壳聚糖衍生物a-67(0.89g),根据其核磁氢谱图,可以计算出化合物88的修饰度大约为12.5%。
[0585]
实施例六十八:组分a-68的合成
[0586][0587]
(1)化合物89的合成:按实施例52的方法,采用常规化学手段制备化合物89。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.76(s,2h),4.42(m,1h),4.13(t,j= 6.1hz,2h),3.99(s,3h),3.95(s,3h),3.43(d,j=5.6,2h),3.04(t,j=7.2hz,2h),1.42(s, 9h).ms(esi):[m+h]523.0731.
[0588]
(2)组分a-68的合成:将peg-4oh(1g,0.05mmol)溶于无水乙腈中,加入k2co
3 (55.3mg,0.4mmol)搅拌30min后,加入化合物89(0.20g,0.4mmol)于室温下继续反应24h。反应结束后,将大部分溶剂除掉,在乙醚中重沉淀,并多次洗涤,抽滤烘干后即可得到光敏聚乙二醇衍生物a-68(0.85g),根据核磁氢谱图,可计算出化合物89的修饰度大约为95%。
[0589]
实施例六十九:组分a-69的合成
[0590][0591]
(1)化合物90的合成:将化合物89(0.5g,1.29mmol)和乙二醇(0.24g,3.87mmol) 溶于无水乙腈中,加入k2co3(0.5g,3.87mmol)做碱,回流过夜反应。待反应结束后,减压旋蒸掉溶剂,过柱纯化,即可得到化合物90(0.34g,72%)。
[0592]
(2)化合物91的合成:将化合物90(0.64g,1.72mmol)和三乙胺(0.34g,3.44mmol) 溶于干燥的二氯甲烷中,冰浴条件下,甲基丙烯酰氯(0.27g,2.58mmol)慢慢逐滴加入到上述溶液中,滴加完后室温条件下过夜反应。反应结束后,减压旋蒸掉溶剂,过柱纯化即可得到化合物91(0.49g,65%)。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h), 6.25(s,1h),5.68(s,1h),4.76(s,2h),4.42(m,1h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.95 (s,3h),3.79(t,j=6.1hz,2h),3.70(t,j=7.2hz,2h),3.56(t,j=7.2hz,2h),3.43(d,j= 5.6,2h),1.87(s,3h),1.42(s,9h).ms(esi):[m+h]573.2125.
[0593]
(3)组分a-69的合成:称取化合物91(0.28g,0.63mmol)、共单体peg-ma(0.882 g,2.52mmol)和引发剂偶氮二异丁腈(11mg)加入到史莱克管中,并加入无水thf溶解,经过多次冷冻-抽真空循环操作处理后,将该反应体系于75℃条件下反应24h。反应结束后,将反应液倒入冷的乙醚中多次重沉淀纯化,即可得到光敏共聚物衍生物a-69(0.86 g)。根据核磁氢谱图,可计算出化合物91在共聚物中的含量大约为15.3%。根据gpc测得合成高分子的分子量在25kda左右,根据投料比计算可得n为12,x为10,y为40。
[0594]
实施例七十:组分a-70的合成
[0595][0596]
(1)化合物92的合成:按参考文献takahiro muraoka.;honggang cui.;samuel i.stupp. j.am.chem.soc.2008,130,2946.公开的方法进行合成。1h nmr(400mhz,cdcl3):δ= 7.71(s,1h),7.22(s,1h),4.35(s,2h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.32(dd,j=11.6, 5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h).ms(esi): [m+h]327.1617.
[0597]
(2)组分a-70的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 92(65mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-70(1.8g),根据核磁氢谱图,可计算出化合物92的标记率大约为3.26%。
[0598]
实施例七十一:组分a-71的合成
[0599][0600]
(1)化合物93的合成:按参考文献takahiro muraoka.;honggang cui.;samuel i.stupp. j.am.chem.soc.2008,130,2946.公开的方法进行合成。
[0601]
(2)化合物94的合成:将化合物93(15.4g,36.24mmol)溶于100ml甲醇中,胺基乙酸甲酯(7.0g,78.65mmol)溶于70ml甲醇和naoh(2m,50ml)水溶液中,然后逐滴加入上述溶液中,室温下搅拌反应30min后,在0℃下缓慢加入nabh4(12g, 317.2mmol)。反应2h后,旋蒸除去溶剂,然后用2m hcl调节ph至5左右,析出白色固体,用乙醚多次洗涤,得到的粗产物用乙醚重沉淀,即化合物94(17.5g,97%)。1h nmr (400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.55(s,2h),4.13(t,j=6.1hz,2h),3.99(s, 3h),3.95(s,3h),3.74(s,2h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t, j=7.2hz,2h),2.26-2.17(m,2h),1.42(s,9h).ms(esi):[m+h]499.2442.
[0602]
(3)化合物95的合成:将化合物94(15g,30mmol)溶于二氯甲烷/三氟乙酸(3:1) 的混合溶液,室温下搅拌反应30min,然后旋蒸除去溶剂,得到的粗产物用乙醚重沉淀,即化合物95(11.4g,95%)。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.55 (s,2h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.95(s,3h),3.74(s,2h),3.32(dd,j=11.6,5.7 hz,
2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h).ms(esi):[m+h] 399.1823.
[0603]
(4)组分a-71的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物95(80mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-71(1.87g),根据核磁氢谱图,可计算出化合物95的标记率大约为3.42%。
[0604]
实施例七十二:组分a-72的合成
[0605][0606]
(1)化合物96的合成:按参考文献james f.cameron.;jean m.j.frechet.j.am.chem. soc.1991,113,4303.公开的方法进行合成。1h nmr(400mhz,cdcl3):δ=7.71(s,1h), 7.22(s,1h),4.75(m,1h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.95(s,3h),3.74(s,2h),3.32 (dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h), 1.33(d,j=6.9hz,3h).ms(esi):[m+h]413.2041.
[0607]
(2)组分a-72的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 96(82mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-72(1.84g),根据核磁氢谱图,可计算出化合物96的标记率大约为3.21%。
[0608]
实施例七十三:组分a-73的合成
[0609][0610]
(1)化合物97的合成:按参考文献jack e.baldwin.;adrian w.mcconnaughie.;sung bo shin.tetrahedron.1990,46,6879.公开的方法进行合成。1h nmr(400mhz,cdcl3):δ= 8.02-7.23(m,5h),7.71(s,1h),7.22(s,1h),4.75(m,1h),4.13(t,j=6.1hz,2h),3.99
(s, 3h),3.95(s,3h),3.74(s,2h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t, j=7.2hz,2h),2.26-2.17(m,2h).ms(esi):[m+h]475.2125.
[0611]
(2)组分a-73的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml 0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 97(95mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-73(1.78g),根据核磁氢谱图,可计算出化合物97的标记率大约为3.14%。
[0612]
实施例七十四:组分a-74的合成
[0613][0614]
(1)化合物98的合成:按参考文献pauloehrl,t.;delaittre,g.;bruns,m.;meiβler,m.;h.g.;bastmeyer,m.;barner-kowollik,c.angew.chem.int.ed.2012,51,9181.公开的方法进行合成。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.55(s,2h), 4.13(t,j=6.1hz,2h),3.99(s,3h),3.90-3.80(m,1h),3.63-3.52(m,1h),3.32(dd,j=11.6, 5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h),2.00-1.34 (m,6h).ms(esi):[m+h]411.2231.
[0615]
(2)组分a-74的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 98(82mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-74(1.88g),根据核磁氢谱图,可计算出化合物98的标记率大约为3.38%。
[0616]
实施例七十五:组分a-75的合成
[0617][0618]
(1)化合物99的合成:按参考文献patchornik abraham.;amit b.;woodward r.b.j. am.chem.soc.1970,92,6333.公开的方法进行合成。1h nmr(400mhz,cdcl3):δ=8.02
ꢀ-
7.23(m,5h),7.71(s,1h),7.22(s,1h),4.55(m,1h),4.13(t,j=6.1hz,2h),3.99(s,
3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m, 2h),1.48(d,j=6.9hz,3h).ms(esi):[m+h]456.2036.
[0627]
(2)组分a-77的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 101(91mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-77(1.82g),根据核磁氢谱图,可计算出化合物101的标记率大约为3.21%。
[0628]
实施例七十八:组分a-78的合成
[0629][0630]
(1)化合物102的合成:按实施例71的方法,采用常规化学手段制备化合物102。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),4.55(s,2h),4.13(t,j=6.1hz,2h),3.99(s, 3h),3.95(s,3h),3.74(s,2h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t, j=7.2hz,2h),2.26-2.17(m,2h).ms(esi):[m+h]433.1426.
[0631]
(2)组分a-78的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 102(86mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-78(1.74g),根据核磁氢谱图,可计算出化合物102的标记率大约为3.09%。
[0632]
实施例七十九:组分a-79的合成
[0633][0634]
(1)化合物103的合成:按实施例71的方法,采用常规化学手段制备化合物103。1h nmr(400mhz,cdcl3):δ=8.02-7.23(m,5h),7.71(s,1h),4.55(s,2h),4.13(t,j=6.1 hz,2h),3.99(s,3h),3.95(s,3h),3.74(s,2h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9 hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h).ms(esi):[m+h]475.2138.
[0635]
(2)组分a-79的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 103(95mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-79(1.76g),根据核磁氢谱图,可计算出化合物103的标记率大约为2.45%。
[0636]
实施例八十:组分a-80的合成
[0637][0638]
(1)化合物104的合成:按参考文献grazyna groszek.;agnieszka nowak-krol.;barbara filipek.eur.j.med.chem.2009,44,5103.公开的方法进行合成。1h nmr(400mhz, cdcl3):δ=8.04(s,1h),7.42(s,1h),4.55(s,2h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.95 (s,3h),3.74(s,2h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz, 2h),2.26-2.17(m,2h).ms(esi):[m+h]399.1832.
[0639]
(2)组分a-80的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 104(80mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-80(1.86g),根据核磁氢谱图,可计算出化合物104的标记率大约为3.32%。
[0640]
实施例八十一:组分a-81的合成
[0641][0642]
(1)化合物105的合成:按参考文献thomas f.greene.;shu wang.;mary j.meegan. j.med.chem.2016,59,90.公开的方法进行合成。1h nmr(400mhz,cdcl3):δ=7.95(s, 1h),7.12(s,1h),4.55(s,2h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.95(s,3h),3.74(s,2h), 3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m, 2h).ms(esi):[m+h]399.1832.
[0643]
(2)组分a-81的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml
0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 105(80mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-81(1.89g),根据核磁氢谱图,可计算出化合物105的标记率大约为3.28%。
[0644]
实施例八十二:组分a-82的合成
[0645][0646]
(1)化合物106的合成:按参考文献yu-shan.;mohane selvaraj coumar.;hsing-pang hsieh.j.med.chem.2009,52,4941.公开的方法进行合成。1h nmr(400mhz,cdcl3):δ= 7.64(s,1h),7.02(s,1h),4.55(s,2h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.95(s,3h),3.74 (s,2h),3.32(dd,j=11.6,5.7hz,2h),2.82(t,j=5.9hz,2h),2.44(t,j=7.2hz,2h),2.26
-ꢀ
2.17(m,2h).ms(esi):[m+h]399.1832.
[0647]
(2)组分a-82的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml 0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 106(80mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-82(1.91g),根据核磁氢谱图,可计算出化合物106的标记率大约为3.26%。
[0648]
实施例八十三:组分a-83的合成
[0649][0650]
组分a-83的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml 0.01mol/l2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取nnb混合物(化合物92/化合物95,60mg,质量比1:1)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6-二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于 3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物a-83(1.89g),根据核磁氢谱图,可计算出nnb混合物(化合物92/化合物95)的标记率大约为3.42%。
nmr(400mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.55(s,2h),4.13(t,j=6.1hz, 2h),3.99(s,3h),3.95(s,3h),3.74(s,2h),3.04(t,j=7.2hz,2h).ms(esi):[m+h] 377.0346.
[0661]
(2)组分a-86的合成:将peg-4oh(1g,0.05mmol)溶于无水乙腈中,加入k2co
3 (55.3mg,0.4mmol)搅拌30min后,加入化合物108(0.15g,0.4mmol)于室温下继续反应24h。反应结束后,将大部分溶剂除掉,在乙醚中重沉淀,并多次洗涤,抽滤烘干后即可得到光敏聚乙二醇衍生物a-86(0.93g),根据核磁氢谱图,可计算出化合物108 的修饰度大约为95%。
[0662]
实施例八十七:组分a-87的合成
[0663][0664]
(1)化合物109的合成:将化合物108(0.5g,1.29mmol)和乙二醇(0.24g,3.87 mmol)溶于无水乙腈中,加入k2co3(0.5g,3.87mmol)做碱,回流过夜反应。待反应结束后,减压旋蒸掉溶剂,过柱纯化,即可得到化合物109(0.34g,72%)。1h nmr(400 mhz,cdcl3):δ=7.71(s,1h),7.22(s,1h),4.55(s,2h),4.13(t,j=6.1hz,2h),3.99(s,3h), 3.95(s,3h),3.79(t,j=6.1hz,2h),3.74(s,2h),3.70(t,j=7.2hz,2h),3.56(t,j=7.2hz, 2h).ms(esi):[m+h]359.1462.
[0665]
(2)化合物110的合成:将化合物109(0.64g,1.72mmol)和三乙胺(0.34g,3.44 mmol)溶于干燥的二氯甲烷中,冰浴条件下,甲基丙烯酰氯(0.27g,2.58mmol)慢慢逐滴加入到上述溶液中,滴加完后室温条件下过夜反应。反应结束后,减压旋蒸掉溶剂,过柱纯化即可得到化合物110(0.49g,65%)。1h nmr(400mhz,cdcl3):δ=7.71(s,1h), 7.22(s,1h),6.25(s,1h),5.68(s,1h),4.55(s,2h),4.13(t,j=6.1hz,2h),3.99(s,3h),3.95 (s,3h),3.79(t,j=6.1hz,2h),3.74(s,2h),3.70(t,j=7.2hz,2h),3.56(t,j=7.2hz,2h), 1.87(s,3h).ms(esi):[m+h]427.1725.
[0666]
(3)组分a-87的合成:称取化合物110(0.28g,0.63mmol)、共单体peg-ma(0.882 g,2.52mmol)和引发剂偶氮二异丁腈(11mg)加入到史莱克管中,并加入无水thf溶解,经过多次冷冻-抽真空循环操作处理后,将该反应体系于75℃条件下反应24h。反应结束后,将反应液倒入冷的乙醚中多次重沉淀纯化,即可得到光敏共聚物衍生物a-87(0.85 g)。根据核磁氢谱图,可计算出化合物110在共聚物中的含量大约为14.6%。根据gpc 测得合成高分子的分子量在25kda左右,根据投料比计算可得n为12,x为10,y为40。
[0667]
实施例八十八:组分a-88的合成
[0668][0669]
(1)化合物111的合成:将香草醛(25g,165mmol)、和碳酸钾(11.4g,83mmol) 溶解在200ml丙酮中,逐滴加入溴化苄(21.2g,181mmol),在90℃条件下回流反应 8h。反应结束后,将反应体系降至室温,然后减压蒸馏除去丙酮,加入100ml水,用乙酸乙酯萃取三次,合并有机相,用无水硫酸钠干燥,旋干有机溶剂得到无色液体。然后用 100ml乙醇重结晶,得到白色针状产物,即化合物111(36.2g,91%)。1h nmr(400mhz, cdcl3):δ=9.83(s,1h),7.39(ddd,j=24.2,20.7,7.4hz,7h),6.98(d,j=8.2hz,1h),5.24(s, 2h),3.94(d,j=0.9hz,3h).ms(esi):[m+na]265.0824.
[0670]
(2)化合物112的合成:将化合物111(10g,41.3mmol)溶解在50ml乙酸酐中,然后在冰浴条件下逐渐滴加50ml的硝酸(65%)。滴加完后,撤去冰浴,在室温条件反应 30min,反应结束后,然后将反应体系缓慢倒入600ml冰水中,有黄色固体析出,减压过滤得到黄色固体,然后用乙醇重结晶,得到黄色针状产物,即化合物112(9.72g,82%)。1h nmr(400mhz,cdcl3):δ=10.42(s,1h),7.67(s,1h),7.43-7.39(m,3h),7.37(d,j=7.0 hz,1h),5.26(s,2h),4.01(s,3h).ms(esi):[m+na]310.0689.
[0671]
(3)化合物113的合成:将化合物112(9g,31.3mmol)溶解在200ml甲醇中,冰浴条件下缓慢加入硼氢化钠(2.37g,62.6mmol),加完后撤去冰浴,室温条件下反应30min,反应结束后,加入2mol/l的盐酸调节反应的ph=7,然后减压蒸馏旋去甲醇,加入100ml 水,用乙酸乙酯萃取三次,合并有机相,并用无水硫酸钠干燥,旋干溶剂即可得到黄色固体产物,再用乙醇进行重结晶,得到黄色固体产物,即化合物113(9.06g,92%)。1h nmr (400mhz,cdcl3):δ=7.77(s,1h),7.49-7.42(m,2h),7.40(dd,j=8.1,6.4hz,3h),7.18(s, 1h),5.20(s,
2h),4.95(s,2h),4.00(s,3h).ms(esi):[m+na]312.0834.
[0672]
(4)化合物114的合成:将化合物113(3g,10.4mmol)溶解到100ml干燥的四氢呋喃中,换气三次,在冰浴条件下同时加入三苯基膦(4.08g,15.6mmol)和四溴化碳(5.16g, 15.6mmol),然后撤去冰浴,室温条件下反应2h,反应结束后,加入6ml水猝灭反应体系,然后减压旋干四氢呋喃,先用饱和食盐水和乙酸乙酯萃取两次,再用水和乙酸乙酯萃取三次,合并有机相,用无水硫酸钠干燥有机相,减压旋干溶剂,干法上样柱层析分离 (pe:ch2cl2=4:1),可得到黄色粉末,即化合物114(3.09g,85%)。1h nmr(400mhz, cdcl3):δ=7.71(s,1h),7.46-7.41(m,2h),7.40-7.30(m,3h),6.93(s,1h),5.17-5.13(m,2h), 4.8-4.79(m,2h),3.95(s,3h),1.42(s,9h).ms(esi):[m+na]374.0003.
[0673]
(5)化合物115的合成:将化合物114(3g,8.5mmol)溶解到120ml丙酮中,换气三次,在氩气保护下加入l-半胱氨酸甲酯盐酸盐(2.9g,17mmol)和氢氧化钠(0.85g, 21.25mmol),再换气三次,室温条件下反应2h,反应结束后,向反应体系中加入4mol/l 的盐酸调节体系ph=7,减压旋干丙酮,先用饱和食盐水和乙酸乙酯萃取三次,再用水和乙酸乙酯萃取三次,合并有机相,用无水硫酸钠干燥。干法上样柱层析分离 (ch2cl2:ch3oh=100:3),可得到黄色固体,即化合物115(2.71g,78%)。1h nmr(400 mhz,cdcl3):δ=7.71(s,1h),7.45(d,j=7.0hz,2h),7.39(t,j=7.2hz,3h),6.95(s,1h), 5.18(s,2h),4.13(q,j=13.6hz,2h),3.98(s,3h),3.73(s,3h),3.65(m,1h),2.91(dd,j= 13.7,4.6hz,1h),2.75(dd,j=13.6,7.5hz,1h).ms(esi):[m+h]407.1277.
[0674]
(6)化合物116的合成:将三缩四乙二醇(22g,113.2mmol)加入到干燥的四氢呋喃中,加入金属钠(40mg,1.74mmol),有气泡产生,待钠完全溶解后,加入丙烯酸叔丁酯(8g,62.4mmol),室温下反应20h,反应结束后,用1mol/l的盐酸调节体系的ph=7,减压旋干四氢呋喃,用饱和食盐水和乙酸乙酯萃取三次,再用水和乙酸乙酯萃取三次,合并有机相,用无水硫酸钠干燥,减压旋干溶剂,无需进一步提纯,得到无色油状液体,即化合物116(16.0g,80%)。1h nmr(400mhz,cdcl3):δ=3.78-3.69(m,4h),3.69-3.54(m, 14h),2.52(dd,j=4.3,2.1hz,2h),1.45(s,9h).ms(esi):[m+na]345.1872.
[0675]
(7)化合物117的合成:将化合物116(10g,31.2mmol)溶于干燥的二氯甲烷中,加入干燥的三乙胺(5.2mml,37.4mmol),然后将对甲基苯磺酰氯(8.9g,46.8mmol) 溶解于40ml干燥的二氯甲烷中,冰浴条件下逐渐滴加到上述反应体系中,滴加完后,撤去冰浴,室温下反应6h。反应结束后,直接向体系中加入200ml水,用二氯甲烷萃取三次,合并有机相并用无水硫酸钠干燥,旋干溶剂,柱层析分离(ch2cl2:ch3oh=50:1),得到淡黄色油状液体,即化合物117(12.6g,85%)。1h nmr(400mhz,cdcl3):δ=7.79-7.74 (m,2h),7.32(d,j=8.5hz,2h),4.21-3.90(m,2h),3.66(dd,j=5.7,2.8hz,4h),3.62-3.35 (m,12h),2.47(dd,j=8.3,4.8hz,2h),2.42(d,j=3.2hz,3h),1.42(d,j=3.4hz,9h).ms (esi):[m+na]499.1964.
[0676]
(8)化合物118的合成:将化合物117(10g,21.0mmol)和溴化锂(4.8g,31.5mmol) 溶解到30ml n,n-二甲基甲酰胺中,加热到80℃反应1h,反应结束后,减压旋干n,n
-ꢀ
二甲基甲酰胺,用水和二氯甲烷萃取三次,合并有机相并用无水硫酸钠干燥,减压旋干溶剂,柱层析分离(ch2cl2),得到淡黄色液体,即化合物118(7.3g,90%)。1h nmr(400 mhz,cdcl3):δ=3.72(t,j=6.3hz,2h),3.62(t,j=6.6hz,2h),3.58(dd,j=2.6,1.5hz,8h), 3.54(d,j=
2.2hz,4h),3.39(t,j=6.3hz,2h),2.42(t,j=6.6hz,2h),1.36(s,9h).ms (esi):[m+na]409.1005.
[0677]
(9)化合物119的合成:将化合物118(5g,13.0mmol)溶解到30ml干燥的二氯甲烷中,加入10ml三氟乙酸,室温下反应30min,反应结束后,减压旋干溶剂,再分别用二氯甲烷和乙酸乙酯溶解产物,再减压旋干溶剂,以达到完全旋干三氟乙酸的目的,无需进一步纯化,得到黄色油状液体,即化合物119(3.9g,92%)。1h nmr(400mhz,cdcl3): δ=3.72(t,j=6.3hz,2h),3.67(t,j=6.3hz,2h),3.58(dd,j=4.1,1.7hz,4h),3.57(s,4h), 3.55(s,4h),3.39(t,j=6.3hz,2h),2.54(t,j=6.3hz,2h).ms(esi):[m+na]353.0414.
[0678]
(10)化合物120的合成:将化合物115(2.0g,4.9mmol)和化合物119(2.0g,5.9mmol) 溶于40ml干燥的二氯甲烷中,加入六氟磷酸苯并三唑-1-基-氧基三吡咯烷基(5.1g, 9.8mmol)和干燥的三乙胺(1.4ml,9.8mmol),室温下反应1h,反应结束后,向体系中加入100ml水,用二氯甲烷和水萃取三次,合并有机相,用无水硫酸钠干燥,减压旋干溶剂,干法上样柱层析分离(ch2cl2:ch3oh=100:3),得到黄色液体,即化合物120 (2.2g,62%)。1h nmr(400mhz,cdcl3):δ=7.71(s,1h),7.45(d,j=7.0hz,2h),7.39(t, j=7.2hz,3h),6.95(s,1h),5.18(s,2h),4.42(m,1h),4.13(q,j=13.6hz,2h),3.98(s,3h), 3.73(s,3h),3.68-3.63(m,2h),3.62-3.55(m,4h),3.58-3.53(m,12h),3.37(t,j=6.3hz, 2h),2.43(t,j=5.8hz,2h).ms(esi):[m+na]741.1529.
[0679]
(11)化合物121的合成:将化合物120(2g,2.8mmol)溶于20ml的三氟乙酸中, 45℃下反应8h,反应结束后,减压旋干三氟乙酸,用二氯甲烷和水萃取三次,合并有机相并用无水硫酸钠干燥,减压旋干溶剂,柱层析分离(ch2cl2:ch3oh=25:1),得到黄色液体,即化合物121(1.4g,82%)。1h nmr(400mhz,cdcl3):δ=7.60(s,1h),6.79(s,1h), 4.73-4.66(m,1h),3.99(d,j=12.9hz,2h),3.97(s,3h),3.73(s,3h),3.70(d,j=6.3hz,2h), 3.62-3.55(m,4h),3.58-3.53(m,12h),3.37(t,j=6.3hz,2h),2.43(t,j=5.8hz,2h).ms (esi):[m+na]651.1026.
[0680]
(12)化合物122的合成:将化合物121(0.5g,0.8mmol)溶于400ml丙酮中,加入(0.2g,1.6mmol)碳酸钾,于75℃下回流反应4h,反应结束后,减压过滤除掉不溶物,然后旋干丙酮,柱层析分离(ch2cl2:ch3oh=25:1),得到黄色固体,即化合物122 (0.27g,61%)。1h nmr(400mhz,dmso):δ=7.71(s,1h),7.17(s,1h),4.76(s,2h),4.42 (m,1h),3.97(s,3h),3.73(s,3h),3.72(d,j=6.3hz,2h),3.62-3.55(m,4h),3.52-3.39(m, 14h),2.49-2.35(m,2h).ms(esi):[m+na]569.1782.
[0681]
(13)化合物123的合成:将化合物122(0.2g,3.7mmol)溶于20ml无水乙二胺中,室温下反应6h,反应结束后,减压旋干乙二胺,干法上样柱层析分离(ch2cl2:ch3oh: 三乙胺=100:8:0.5),得到黄色粉末,即化合物123(0.19g,89%)。1h nmr(400mhz,dmso): δ=7.71(s,1h),7.17(s,1h),4.76(s,2h),4.42(m,1h),3.97(s,3h),3.72(d,j=6.3hz,2h), 3.62-3.55(m,2h),3.52-3.39(m,16h),2.86(d,j=7.6hz,2h),2.76(d,j=7.6hz,2h),2.49
ꢀ-
2.35(m,2h).ms(esi):[m+na]597.2211.
2.76(d,j=7.6hz,2h),2.49-2.35(m,2h).ms(esi):[m+na]558.2725.
[0691]
(2)组分a-90的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 125(111mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-90(1.87g),根据核磁氢谱图,可计算出化合物125的标记率大约为3.27%。
[0692]
实施例九十一:组分a-91的合成
[0693][0694]
(1)化合物126的合成:按实施例88的方法,采用化学手段制备化合物126。1h nmr (400mhz,dmso):δ=7.71(s,1h),7.17(s,1h),5.16(m,1h),4.42(m,1h),3.97(s,3h),3.72 (d,j=6.3hz,2h),3.62-3.55(m,2h),3.52-3.39(m,16h),2.86(d,j=7.6hz,2h),2.76(d,j =7.6hz,2h),2.49-2.35(m,2h),1.33(d,j=6.9hz,3h).ms(esi):[m+na]589.2517.
[0695]
(2)组分a-91的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 126(118mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-91(1.73g),根据核磁氢谱图,可计算出化合物126的标记率大约为3.14%。
[0696]
实施例九十二:组分a-92的合成
[0697][0698]
(1)化合物127的合成:按实施例88的方法,采用化学手段制备化合物127。1h nmr (400mhz,dmso):δ=8.02-7.23(m,5h),7.71(s,1h),7.17(s,1h),5.34(m,1h),4.42(m, 1h),3.97(s,3h),3.72(d,j=6.3hz,2h),3.62-3.55(m,2h),3.52-3.39(m,16h),2.86(d,j= 7.6hz,2h),2.76(d,j=7.6hz,2h),2.49-2.35(m,2h).ms(esi):[m+na]651.2761.
[0699]
(2)组分a-92的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml 0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 127(130mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔
1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-92(1.78g),根据核磁氢谱图,可计算出化合物127的标记率大约为3.09%。
[0700]
实施例九十三:组分a-93的合成
[0701][0702]
(1)化合物128的合成:按实施例88的方法,采用常规化学手段制备化合物128。1h nmr(400mhz,dmso):δ=7.71(s,1h),7.17(s,1h),5.82(m,1h),4.76(s,2h),3.97(s, 3h),3.62-3.55(m,2h),3.52-3.39(m,16h),2.86(d,j=7.6hz,2h),2.76(d,j=7.6hz,2h), 2.49-2.35(m,2h).ms(esi):[m+na]589.2143.
[0703]
(2)组分a-93的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 128(118mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-93(1.73g),根据核磁氢谱图,可计算出化合物128的标记率大约为3.15%。
[0704]
实施例九十四:组分a-94的合成
[0705][0706]
(1)化合物129的合成:按实施例88的方法,采用常规化学手段制备化合物129。1h nmr(400mhz,dmso):δ=7.71(s,1h),7.17(s,1h),4.76(s,2h),4.13(t,j=7.2hz,2h), 3.62-3.55(m,2h),3.52-3.39(m,16h),2.86(d,j=7.6hz,2h),2.76(d,j=7.6hz,2h),2.49
ꢀ-
2.35(m,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h).ms(esi):[m+na]575.2332.
[0707]
(2)组分a-94的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 129(115mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-94(1.84g),根据核磁氢谱图,可计算出化合物129的标记率大约为2.47%。
[0708]
实施例九十五:组分a-95的合成
[0709][0710]
(1)化合物130的合成:按实施例88的方法,采用常规化学手段制备化合物130。1h nmr(400mhz,dmso):δ=7.71(s,1h),7.17(s,1h),4.76(s,2h),4.13(t,j=7.2hz,2h), 3.62-3.55(m,2h),3.52-3.39(m,16h),2.86(d,j=7.6hz,2h),2.76(d,j=7.6hz,2h),2.69
ꢀ-
2.55(m,2h),2.44(t,j=7.2hz,2h),2.26-2.17(m,2h).ms(esi):[m+na]576.2242.
[0711]
(2)组分a-95的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物130(115mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-95(1.75g),根据核磁氢谱图,可计算出化合物130的标记率大约为3.07%。
[0712]
实施例九十六:组分a-96的合成
[0713][0714]
(1)化合物131的合成:按实施例88的方法,采用常规化学手段制备化合物131。1h nmr(400mhz,dmso):δ=7.71(s,1h),4.76(s,2h),4.42(m,1h),3.97(s,3h),3.72(d,j =6.3hz,2h),3.62-3.55(m,2h),3.52-3.39(m,16h),2.86(d,j=7.6hz,2h),2.76(d,j= 7.6hz,2h),2.49-2.35(m,2h).ms(esi):[m+na]609.2053.
[0715]
(2)组分a-96的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 131(122mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-96(1.76g),根据核磁氢谱图,可计算出化合物131的标记率大约为3.24%。
[0716]
实施例九十七:组分a-97的合成
[0717]
[0718]
(1)化合物132的合成:按实施例88的方法,采用常规化学手段制备化合物132。1h nmr(400mhz,dmso):δ=8.02-7.23(m,5h),7.71(s,1h),4.76(s,2h),4.42(m, 1h),3.97(s,3h),3.72(d,j=6.3hz,2h),3.62-3.55(m,2h),3.52-3.39(m,16h),2.86(d,j=7.6hz,2h),2.76(d,j=7.6hz,2h),2.49-2.35(m,2h).ms(esi):[m+na]651.2722.
[0719]
(2)组分a-97的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 132(130mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-97(1.82g),根据核磁氢谱图,可计算出化合物132的标记率大约为3.05%。
[0720]
实施例九十八:组分a-98的合成
[0721][0722]
(1)化合物133的合成:按实施例88的方法,采用常规化学手段制备化合物133。1h nmr(400mhz,dmso):δ=8.11(m,1h),7.27(m,1h),4.76(s,2h),4.42(m,1h),3.97(s, 3h),3.72(d,j=6.3hz,2h),3.62-3.55(m,2h),3.52-3.39(m,12h),2.86(d,j=7.6hz,2h), 2.76(d,j=7.6hz,2h),2.49-2.35(m,2h).ms(esi):[m+na]531.2143.
[0723]
(2)组分a-98的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 133(106mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-98(1.78g),根据核磁氢谱图,可计算出化合物133的标记率大约为3.31%。
[0724]
实施例九十九:组分a-99的合成
[0725][0726]
(1)化合物134的合成:按实施例88的方法,采用常规化学手段制备化合物134。1h nmr(400mhz,dmso):δ=7.71(s,1h),7.17(s,1h),4.76(s,2h),4.42(m,1h),3.97(s, 3h),3.72(d,j=6.3hz,2h),3.62-3.55(m,2h),3.52-3.39(m,16h),2.86(d,j=7.6hz,2h), 2.76(d,j=7.6hz,2h),2.49-2.35(m,2h).ms(esi):[m+na]575.2342.
[0727]
(2)组分a-99的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml
0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 134(115mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-99(1.84g),根据核磁氢谱图,可计算出化合物134的标记率大约为3.06%。
[0728]
实施例一百:组分a-100的合成
[0729][0730]
(1)化合物135的合成:按实施例88的方法,采用常规化学手段制备化合物135。1h nmr(400mhz,dmso):δ=7.54(m,1h),7.03(m,1h),4.76(s,2h),4.42(m,1h),3.97(s, 3h),3.72(d,j=6.3hz,2h),3.62-3.55(m,2h),3.52-3.39(m,20h),2.86(d,j=7.6hz,2h), 2.76(d,j=7.6hz,2h),2.49-2.35(m,2h).ms(esi):[m+na]619.2652.
[0731]
(2)组分a-100的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 135(124mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物 a-100(1.84g),根据核磁氢谱图,可计算出化合物135的标记率大约为3.16%。
[0732]
实施例一百零一:组分a-101的合成
[0733][0734]
(1)化合物136的合成:按实施例88的方法,采用常规化学手段制备化合物136。1h nmr(400mhz,dmso):δ=7.71(s,1h),7.17(s,1h),4.76(s,2h),4.42(m,1h),4.13(t,j =7.2hz,2h),3.73(s,3h),3.72(d,j=6.3hz,2h),3.62-3.55(m,2h),3.52-3.39(m,16h), 2.86(d,j=7.6hz,2h),2.76(d,j=7.6hz,2h),2.49-2.35(m,2h),2.44(t,j=7.2hz,2h), 2.26-2.17(m,2h).ms(esi):[m+na]661.2745.
[0735]
(2)组分a-101的合成:将透明质酸hyaluronic acid(2g,340kda)溶于100ml0.01mol/l 2-(n-吗啉)乙磺酸mes缓冲溶液(ph=5.2),搅拌至完全溶解,称取化合物 136(132mg,0.2mmol)溶于10ml二甲基亚砜dmso后加入上述反应液,称取4-(4,6
-ꢀ
二甲氧基三嗪-2-基)-4-甲基吗啉盐酸盐dmtmm(0.4g,1.5mmol)溶于3ml mes缓冲溶液,分三次(每隔1h)加入上述反应液中,35℃下反应24h。然后将反应液倒入透析袋(mwco 7000)中,用去离
nmr(400mhz,dmso):δ=7.71(s,1h),7.17(s,1h),4.76(s,2h),4.42(m,1h),3.97(s, 3h),3.72(d,j=6.3hz,2h),3.62-3.55(m,2h),3.52-3.39(m,16h),2.49-2.35(m,2h).ms (esi):[m+na]533.1845.
[0745]
(2)组分a-104的合成:将1g壳聚糖加入到75ml异丙醇中形成壳聚糖的悬浮液,然后将化合物137(0.2g,0.35mmol)、edc-hcl(0.76g,3.96mmol)和nhs(0.46g, 4.0mmol)依次加入到上述溶液中,室温下搅拌反应48h。反应结束后,先用含氯化钠的稀盐酸溶液(ph=3.5)透析1d,再用纯水透析1d后,冷冻干燥即可得到光敏壳聚糖衍生物a-104(0.82g),根据其核磁氢谱图,可以计算出化合物137的修饰度大约为12.5%。
[0746]
实施例一百零五:组分a-105的合成
[0747][0748]
(1)化合物138的合成:按实施例88的方法,采用常规化学手段制备化合物138。1h nmr(400mhz,dmso):δ=7.71(s,1h),7.17(s,1h),4.76(s,2h),4.42(m,1h),4.13(t,j =6.1hz,2h),3.93(s,3h),3.72(d,j=6.3hz,2h),3.62-3.55(m,2h),3.52-3.39(m,16h), 3.04(t,j=7.2hz,2h),2.49-2.35(m,2h).ms(esi):[m+na]640.1134.
[0749]
(2)组分a-105的合成:将peg-4oh(1g,0.05mmol)溶于无水乙腈中,加入 k2co3(55.3mg,0.4mmol)搅拌30min后,加入化合物138(0.23g,0.4mmol)于室温下继续反应24h。反应结束后,将大部分溶剂除掉,在乙醚中重沉淀,并多次洗涤,抽滤烘干后即可得到光敏聚乙二醇衍生物a-105(0.85g),根据核磁氢谱图,可计算出化合物138的修饰度大约为95.3%。
[0750]
实施例一百零六:组分a-106的合成
[0751][0752]
(1)化合物139的合成:按实施例88的方法,采用常规化学手段制备化合物139。1h nmr(400mhz,dmso):δ=7.71(s,1h),7.17(s,1h),6.25(s,1h),5.68(s,1h),4.76(s, 2h),4.42(m,1h),4.13(t,j=6.1hz,2h),3.93(s,3h),3.79(t,j=6.1hz,2h),3.72(d,j=6.3 hz,2h),3.70(t,j=7.2hz,2h),3.62-3.55(m,2h),3.56(t,j=7.2hz,2h),3.52-3.39(m, 16h),2.49-2.35(m,2h),1.87(s,3h).ms(esi):[m+na]689.2523.
[0753]
(2)组分a-106的合成:称取化合物139(0.28g,0.63mmol)、共单体peg-ma(0.882 g,2.52mmol)和引发剂偶氮二异丁腈(11mg)加入到史莱克管中,并加入无水thf溶解,经过多次冷冻-抽真空循环操作处理后,将该反应体系于75℃条件下反应24h。反应结束后,将反
应液倒入冷的乙醚中多次重沉淀纯化,即可得到光敏共聚物衍生物a-106 (0.85g)。根据核磁氢谱图,可计算出化合物139在共聚物中的含量大约为15.4%。根据 gpc测得合成高分子的分子量在25kda左右,根据投料比计算可得n为12,x为10,y 为40。
[0754]
实施例一百零七:组分a-107的合成
[0755][0756]
组分a-107的合成:将透明质酸hyaluronic acid(1g,48kda)溶于100ml去离子水,冷却至0-4℃,加入4ml甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco 3500)中,用去离子水透析2-3d,冷冻干燥即可得到光敏透明质酸衍生物a-107(0.92g),根据核磁氢谱图,可计算出双键的含量大约为54%。
[0757]
实施例一百零八:组分a-108的合成
[0758][0759]
组分a-108的合成:将羧甲基纤维素carboxymethyl cellulose(1g,90kda)溶于100 ml去离子水,冷却至0-4℃,加入4ml甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco 3500)中,用去离子水透析2-3d,冷冻干燥即可得到光敏羧甲基纤维素衍生物a-108(0.89g),根据核磁氢谱图,可计算出双键的含量大约为43%。
[0760]
实施例一百零九:组分a-109的合成
[0761][0762]
组分a-109的合成:将海藻酸alginate(1g,48kda)溶于100ml去离子水,冷却至0-4℃,加入4ml甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco 3500)中,用去离子水透析2-3d,冷冻干燥即可得到光敏海藻酸衍生物a-109(0.87g),根据核磁氢谱图,可计算出双键的含量大约为57%。
[0763]
实施例一百十:组分a-110的合成
[0764][0765]
组分a-110的合成:将硫酸软骨素chondroitin sulfate(1g)溶于100ml去离子水,冷却至0-4℃,加入4ml甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco 3500)中,用去离子水透析2-3d,冷冻干燥即可得到光敏硫酸软骨素衍生物a-110(0.91g),根据核磁氢谱图,可计算出双键的含量大约为49%。
[0766]
实施例一百十一:组分a-111的合成
[0767][0768]
组分a-111的合成:将葡聚糖dextran(6g,70kda)溶于60ml无水二甲基亚砜dmso 中,加入2ml三乙胺tea,再加入0.56ml丙烯酰氯(溶于10ml二氯甲烷dcm中),反应10h,反应结束后,将反应液倒入乙醇中重沉淀,过滤得到的粗产物重新溶于去离子水中,透析2-3d,冷冻干燥即可得到光敏葡聚糖衍生物a-111(5.8g),根据核磁氢谱图,可计算出双键的含量大约为24%。
[0769]
实施例一百十二:组分a-112的合成
[0770][0771]
组分a-112的合成:将羧甲基壳聚糖carboxymethylchitosan(1g)溶于100ml去离子水,加热至40℃搅拌溶解,加入4ml甲基丙烯酸缩水甘油酯,再加入2ml 5m naoh,反应2-3h后,将反应液倒入透析袋(mwco 3500)中,用去离子水透析2-3d,冷冻干燥即可得到光敏壳聚糖衍生物a-112(0.88g),根据核磁氢谱图,可计算出双键的含量大约为32%。
[0772]
实施例一百十三:组分a-113的合成
[0773][0774]
组分a-113的合成:将明胶gelatin(1g)溶于10ml d-pbs中,加热至50℃搅拌至完全溶解,加入0.5ml甲基丙烯酸酐,反应2-3h,反应后用40ml d-pbs稀释反应液,然后倒入透析袋(mwco 3500)中,用去离子水透析2-3d,冷冻干燥即可得到光敏明胶衍生物a-113(0.93g),根据核磁氢谱图,可计算出双键的含量大约为56%。
[0775]
实施例一百十四:组分a-114的合成
[0776][0777]
组分a-114的合成:将两臂羟基聚乙二醇peg(10kda,10g)溶于干燥的二氯甲烷中,加入三乙胺(0.28ml,2mmol),再缓慢滴加丙烯酰氯(0.18g,2mmol)的二氯甲烷溶液于上述溶液中,搅拌反应12h,然后将反应液倒入透析袋(mwco 3500)中,用去离子水透析2-3d,冷冻干燥即可得到光敏聚乙二醇衍生物a-114(9.8g),根据核磁氢谱图,可计算出双键的含量大约为98%。
[0778]
实施例一百十五:组分a-115的合成
[0779]
[0780]
组分a-115的合成:将四臂羟基聚乙二醇peg(10kda,10g)溶于干燥的二氯甲烷中,加入三乙胺(0.56ml,4mmol),再缓慢滴加丙烯酰氯(0.36g,4mmol)的二氯甲烷溶液于上述溶液中,搅拌反应12h,然后将反应液倒入透析袋(mwco 3500)中,用去离子水透析2-3d,冷冻干燥即可得到光敏聚乙二醇衍生物a-115(9.3g),根据核磁氢谱图,可计算出双键的含量大约为96%。
[0781]
实施例一百十六:组分a-116的合成
[0782][0783]
组分a-116的合成:将组分a-1溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-116(0.91g),根据核磁氢谱图,可计算出双键的含量大约为54%。
[0784]
实施例一百十七:组分a-117的合成
[0785][0786]
组分a-117的合成:将组分a-2溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-117(0.87g),根据核磁氢谱图,可计算出双键的含量大约为51%。
[0787]
实施例一百十八:组分a-118的合成
[0788][0789]
组分a-118的合成:将组分a-8溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-118(0.86g),根据核磁氢谱图,可计算出双键的含量大约为44%。
[0790]
实施例一百十九:组分a-119的合成
[0791][0792]
组分a-119的合成:将组分a-28溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-119(0.85g),根据核磁氢谱图,可计算出双键的含量大约为43%。
[0793]
实施例一百二十:组分a-120的合成
[0794][0795]
组分a-120的合成:将组分a-29溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-120(0.93g),根据核磁氢谱图,可计算出双键的含量大约为55%。
[0796]
实施例一百二十一:组分a-121的合成
[0797][0798]
组分a-121的合成:将组分a-30溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-121(0.85g),根据核磁氢谱图,可计算出双键的含量大约为49%。
[0799]
实施例一百二十二:组分a-122的合成
[0800][0801]
组分a-122的合成:将组分a-37溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏羧甲基纤维素衍生物a-122(0.91g),根据核磁氢谱图,可计算出双键的含量大约为 42%。
[0802]
实施例一百二十三:组分a-123的合成
[0803][0804]
组分a-123的合成:将组分a-43溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏壳聚糖衍生物a-123(0.84g),根据核磁氢谱图,可计算出双键的含量大约为56%。
[0805]
实施例一百二十四:组分a-124的合成
[0806][0807]
组分a-124的合成:将组分a-45溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco
ꢀꢀ
3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏明胶衍生物a-124(0.92g),根据核磁氢谱图,可计算出双键的含量大约为48%。
[0808]
实施例一百二十五:组分a-125的合成
[0809][0810]
组分a-125的合成:将组分a-49溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏聚乙二醇衍生物a-125(0.94g),根据核磁氢谱图,可计算出双键的含量大约为24%。
[0811]
实施例一百二十六:组分a-126的合成
[0812][0813]
组分a-126的合成:将组分a-51溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-126(0.87g),根据核磁氢谱图,可计算出双键的含量大约为46%。
[0814]
实施例一百二十七:组分a-127的合成
[0815][0816]
组分a-127的合成:将组分a-52溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-127(0.85g),根据核磁氢谱图,可计算出双键的含量大约为57%。
[0817]
实施例一百二十八:组分a-128的合成
[0818][0819]
组分a-128的合成:将组分a-53溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-128(0.93g),根据核磁氢谱图,可计算出双键的含量大约为47%。
[0820]
实施例一百二十九:组分a-129的合成
[0821][0822]
组分a-129的合成:将组分a-62溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用
去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-129(0.90g),根据核磁氢谱图,可计算出双键的含量大约为58%。
[0823]
实施例一百三十:组分a-130的合成
[0824][0825]
组分a-130的合成:将组分a-63溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-130(0.89g),根据核磁氢谱图,可计算出双键的含量大约为46%。
[0826]
实施例一百三十一:组分a-131的合成
[0827][0828]
组分a-131的合成:将组分a-64溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-131(0.87g),根据核磁氢谱图,可计算出双键的含量大约为58%。
[0829]
实施例一百三十二:组分a-132的合成
[0830][0831]
组分a-132的合成:将组分a-66溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏羧甲基纤维素衍生物a-132(0.92g),根据核磁氢谱图,可计算出双键的含量大约为 46%。
[0832]
实施例一百三十三:组分a-133的合成
[0833][0834]
组分a-133的合成:将组分a-67溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏壳聚糖衍生物a-133(0.91g),根据核磁氢谱图,可计算出双键的含量大约为47%。
[0835]
实施例一百三十四:组分a-134的合成
[0836][0837]
组分a-134的合成:将组分a-68溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基
丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏聚乙二醇衍生物a-134(0.87g),根据核磁氢谱图,可计算出双键的含量大约为52%。
[0838]
实施例一百三十五:组分a-135的合成
[0839][0840]
组分a-135的合成:将组分a-70溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-135(0.92g),根据核磁氢谱图,可计算出双键的含量大约为47%。
[0841]
实施例一百三十六:组分a-136的合成
[0842][0843]
组分a-136的合成:将组分a-71溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-136(0.86g),根据核磁氢谱图,可计算出双键的含量大约为51%。
[0844]
实施例一百三十七:组分a-137的合成
[0845][0846]
组分a-137的合成:将组分a-72溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-137(0.93g),根据核磁氢谱图,可计算出双键的含量大约为45%。
[0847]
实施例一百三十八:组分a-138的合成
[0848][0849]
组分a-138的合成:将组分a-80溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-138(0.90g),根据核磁氢谱图,可计算出双键的含量大约为48%。
[0850]
实施例一百三十九:组分a-139的合成
[0851]
[0852]
组分a-139的合成:将组分a-81溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-139(0.88g),根据核磁氢谱图,可计算出双键的含量大约为46%。
[0853]
实施例一百四十:组分a-140的合成
[0854][0855]
组分a-140的合成:将组分a-82溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco 3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-140(0.91g),根据核磁氢谱图,可计算出双键的含量大约为57%。
[0856]
实施例一百四十一:组分a-141的合成
[0857][0858]
组分a-141的合成:将组分a-84溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏羧甲基纤维素衍生物a-141(0.92g),根据核磁氢谱图,可计算出双键的含量大约为 44%。
[0859]
实施例一百四十二:组分a-142的合成
[0860][0861]
组分a-142的合成:将组分a-85溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏壳聚糖衍生物a-142(0.87g),根据核磁氢谱图,可计算出双键的含量大约为56%。
[0862]
实施例一百四十三:组分a-143的合成
[0863][0864]
组分a-143的合成:将组分a-86溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏聚乙二醇衍生物a-143(0.91g),根据核磁氢谱图,可计算出双键的含量大约为48%。
[0865]
实施例一百四十四:组分a-144的合成
[0866][0867]
组分a-144的合成:将组分a-88溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-144(0.89g),根据核磁氢谱图,可计算出双键的含量大约为52%。
[0868]
实施例一百四十五:组分a-145的合成
[0869][0870]
组分a-145的合成:将组分a-89溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-145(0.81g),根据核磁氢谱图,可计算出双键的含量大约为43%。
[0871]
实施例一百四十六:组分a-146的合成
[0872][0873]
组分a-146的合成:将组分a-90溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-146(0.84g),根据核磁氢谱图,可计算出双键的含量大约为49%。
[0874]
实施例一百四十七:组分a-147的合成
[0875][0876]
组分a-147的合成:将组分a-91溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-147(0.92g),根据核磁氢谱图,可计算出双键的含量大约为46%。
[0877]
实施例一百四十八:组分a-148的合成
[0878][0879]
组分a-148的合成:将组分a-98溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-148(0.94g),根据核磁氢谱图,可计算出双键的含量大约为56%。
[0880]
实施例一百四十九:组分a-149的合成
[0881][0882]
组分a-149的合成:将组分a-99溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-149(0.87g),根据核磁氢谱图,可计算出双键的含量大约为51%。
[0883]
实施例一百五十:组分a-150的合成
[0884][0885]
组分a-150的合成:将组分a-100溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-150(0.88g),根据核磁氢谱图,可计算出双键的含量大约为47%。
[0886]
实施例一百五十一:组分a-151的合成
[0887][0888]
组分a-151的合成:将组分a-101溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏透明质酸衍生物a-151(0.91g),根据核磁氢谱图,可计算出双键的含量大约为45%。
[0889]
实施例一百五十二:组分a-152的合成
[0890][0891]
组分a-152的合成:将组分a-103溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏羧甲基纤维素衍生物a-152(0.84g),根据核磁氢谱图,可计算出双键的含量大约为 43%。
[0892]
实施例一百五十三:组分a-153的合成
[0893][0894]
组分a-153的合成:将组分a-104溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏壳聚糖衍生物a-153(0.89g),根据核磁氢谱图,可计算出双键的含量大约为50%。
[0895]
实施例一百五十四:组分a-154的合成
[0896][0897]
组分a-154的合成:将组分a-105溶于100ml去离子水,冷却至0-4℃,加入4ml 甲基丙烯酸酐,再缓慢滴加2ml 5m naoh,反应24h,然后将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到既含邻硝基苄基又含双键官能团的光敏聚乙二醇衍生物a-154(0.94g),根据核磁氢谱图,可计算出双键的含量大约为43%。
[0898]
实施例一百五十五:光引发剂-lap的合成
[0899][0900]
lap的合成:将二甲氧基苯膦(3.0g,0.018mol)置于250ml三口烧瓶中,氩气保护,加入2,4,6-三甲基苯甲酰氯(3.2g,0.018mol),室温下搅拌反应18h,然后将溴化锂(6.1g,0.072mol)溶于100ml2-丁酮中,加入上述反应液中,加热至50℃反应 10min后,有沉淀生成,冷却至室温静置4h后,过滤,得到的粗产物用2-丁酮多次洗涤,干燥后即可得白色固体lap(6.0g)。
[0901]
实施例一百五十六:组分c-10的合成
[0902][0903]
组分c-10的合成:将羧甲基纤维素cmc(400mg)溶于50ml蒸馏水中至完全溶解,加入羟基苯并三唑(hobt,153mg)、二联胺(90mg)和1-乙基-(3-二甲基胺基丙基)碳二亚胺盐酸盐(edc-hcl,90mg)加入到上述溶液中室温反应48h后,先用含氯化钠的稀盐酸溶液(ph=3.5)透析1d,再用纯水透析1d后,冷冻干燥即可得到联胺修饰的羧甲基纤维素(410mg)。tbns方法测试联胺的接枝率约为10%。
[0904]
实施例一百五十七:组分c-11的合成
[0905][0906]
组分c-11的合成:将透明质酸ha(400mg)溶于50ml蒸馏水中至完全溶解,加入羟基苯并三唑(hobt,153mg)、碳二酰肼(cdh,90mg)和1-乙基-(3-二甲基胺基丙基)碳二亚胺盐酸盐(edc-hcl,90mg)加入到上述溶液中室温反应48h后,先用含氯化钠的稀盐酸溶液(ph=3.5)透析1d,再用纯水透析1d后,冷冻干燥即可得到ha-cdh (410mg)。tbns方法测试酰肼的接枝率约为10%。
[0907]
实施例一百五十八:组分c-12的合成
[0908][0909]
组分c-12的合成:将透明质酸ha(400mg)溶于50ml蒸馏水中至完全溶解,加入羟基苯并三唑(hobt,153mg)、草酸二酰肼(odh,90mg)和1-乙基-(3-二甲基胺基丙基)碳二亚胺盐酸盐(edc-hcl,90mg)加入到上述溶液中室温反应48h后,先用含氯化钠的稀盐酸溶液(ph=3.5)透析1d,再用纯水透析1d后,冷冻干燥即可得到ha-odh(410mg)。tbns方法测试酰肼的接枝率约为10%。
[0910]
实施例一百五十九:组分c-13的合成
[0911][0912]
组分c-13的合成:将透明质酸ha(400mg)溶于50ml蒸馏水中至完全溶解,加入羟基苯并三唑(hobt,153mg)、己二酸二酰肼(adh,90mg)和1-乙基-(3-二甲基胺基丙基)碳二亚胺盐酸盐(edc-hcl,90mg)加入到上述溶液中室温反应48h后,先用含氯化钠的稀盐酸溶液(ph=3.5)透析1d,再用纯水透析1d后,冷冻干燥即可得到 ha-adh(410mg)。tbns方法测试
酰肼的接枝率约为10%。
[0913]
实施例一百六十:组分c-14的合成
[0914][0915]
组分c-14的合成:称取四臂羟基聚乙二醇(peg-4oh,2g,97.3μmol)和n-羟基邻苯二甲酰亚胺(634.6mg,3.89mmol)溶于干燥的二氯甲烷中,然后在冰浴条件下慢慢加入三苯基膦(1.02g,3.89mmol)并反应约30min。将偶氮二羧酸二异丙酯(765.9μl, 3.89mmol)溶于干燥的二氯甲烷中慢慢滴加到上述溶液中并在室温下反应1d。反应结束后,将n-羟基邻苯二甲酰亚胺修饰的四臂聚乙二醇用乙醚重沉淀。然后将上述物质(0.25 g,11.8μmol)重新溶解于乙腈中,加入一水合肼(22.9μl,473μmol)继续搅拌2h。然后向该混合物溶液中加入二氯甲烷并抽滤。将滤液减压旋蒸除掉溶剂,即可得到羟胺修饰的四臂聚乙二醇(peg-4onh2)。
[0916]
实施例一百六十一:组分c-15的合成
[0917][0918]
组分c-15的合成:称取葡聚糖(dextran,2g,97.3μmol)和n-羟基邻苯二甲酰亚胺(634.6mg,3.89mmol)溶于干燥的二氯甲烷中,然后在冰浴条件下慢慢加入三苯基膦(1.02g,3.89mmol)并反应约30min。将偶氮二羧酸二异丙酯(765.9μl,3.89mmol) 溶于干燥的二氯甲烷中慢慢滴加到上述溶液中并在室温下反应1d。反应结束后,将n
-ꢀ
羟基邻苯二甲酰亚胺修饰的葡聚糖用乙醚重沉淀。然后将上述物质(0.25g,11.8μmol) 重新溶解于乙腈中,加入一水合肼(22.9μl,473μmol)继续搅拌2h。然后向该混合物溶液中加入二氯甲烷并抽滤。将滤液减压旋蒸除掉溶剂,即可得到羟胺修饰的葡聚糖 (dex-onh2)。
[0919]
实施例一百六十二:组分c-18的合成
[0920][0921]
组分c-18的合成:将透明质酸hyaluronic acid(0.5g,48kda)溶于50ml蒸馏水中至完全溶解,加入羟基苯并三唑(hobt,0.2g),1-乙基-(3-二甲基胺基丙基)碳二亚胺盐酸盐(edc-hcl,0.1g),3,3
’-
二硫代双(丙酰肼)(dtp,0.1g),用稀盐酸溶液调节ph至4.75,反应24h,然后加入dtt继续反应5h后,将反应液倒入透析袋(mwco3500)中,用去离子水透析2-3d,冷冻干燥即可得到ha-sh(0.45g),根据核磁氢谱图,可计算出巯基的含量大约为20%。
[0922]
实施例一百六十三:组分c-19的合成
[0923]
[0924]
组分c-19的合成:将羧甲基壳聚糖carboxymethyl chitosan(1g)溶于100ml去离子水,加入n-乙酰基半胱氨酸(1.77g,10mmol),再加入1-乙基-(3-二甲基胺基丙基) 碳二亚胺盐酸盐edc-hcl(1.91g,10mmol),然后用盐酸调节ph至5左右,室温下搅拌反应5h,然后将反应液倒入透析袋(mwco 3500)中,用5mm hcl溶液透析1d,然后用5mm hcl/1%nacl溶液透析1d,最后用1mm hcl溶液透析1d,冷冻干燥即可得到cmch-sh(0.9g),根据核磁氢谱图,可计算出巯基的含量大约为10%。
[0925]
实施例一百六十四:组分c-20的合成
[0926][0927]
组分c-20的合成:将40kda葡聚糖dextran(12g,0.3mmol)溶于50ml dmso 中至完全溶解,加入3-巯基丙酸(636.8mg,6.0mmol),1,3-二环己基碳二亚胺(910.7mg, 9.0mmol),4-(二甲胺基)吡啶(1099.5mg,9.0mmol),于室温下反应48h,然后在丙酮中重沉淀,将粗产物溶于水后倒入透析袋(mwco 3500)中,用去离子水透析2-3d,冷冻干燥即可得到dex-sh(11.5g),根据核磁氢谱图,可计算出巯基的含量大约为20%。
[0928]
实施例一百六十五:组分c-21的合成
[0929][0930]
组分c-21的合成:将肝素heparin(0.5g,12kda)溶于50ml蒸馏水中至完全溶解,加入羟基苯并三唑(hobt,0.2g),1-乙基-(3-二甲基胺基丙基)碳二亚胺盐酸盐 (edc-hcl,0.1g),巯基乙胺(0.1g),用稀盐酸溶液调节ph至5-6,反应24h后,将反应液倒入透析袋(mwco 3500)中,用去离子水透析2-3d,冷冻干燥即可得到hep-sh (0.45g),根据核磁氢谱图,可计算出巯基的含量大约为20%。
[0931]
实施例一百六十六:光交联方法制备水凝胶
[0932]
按照本发明方法,于37℃下操作,制得不同的水凝胶前体溶液,如表1所示。
[0933]
表1
[0934]
[0935]
将上述不同凝胶溶液分别在365或395nm(20mw/cm2)条件下照射一定时间,即可得到不同化学组成的水凝胶。不同的凝胶材料具有不同的生物效应,可以根据不同的应用针对性地选择凝胶材料的组成。
[0936]
注:组分a
…
为组分a-2~a-153;组分a
……
为组分a-1~a-154;组分c
…
为组分 c-1~c-21。
[0937]
表1中1-20wt%为水凝胶前体溶液优选的质量浓度范围。
[0938]
实施例一百六十七:光交联水凝胶流变测试
[0939]
流变分析采用haake mars流变仪,在37℃的测试平台上进行流变测试。本实例研究了紫外光照时间、光照强度和高分子衍生物的质量浓度对水凝胶成胶时间和储存模量的影响。图1为实施例一制备的组分a-1(即为ha-nb)、实施例一百零七制备的组分a-107(即为hama)、组分c-4(即为明胶)、实施例一百五十五制备的组分 b-2(即为lap);实施例八十八制备的组分a-88(即为ha-cnb)、实施例一百四十四制备的组分a-144(即为ha-cnb-ma)、实施例一百五十五制备的组分b-2(即为lap)配制的水凝胶前体溶液在光照下的成胶曲线(流变测试中,g
’
为储存模量,g”为损耗模量,当g
’
超过g”时即为凝胶点),因此,无论在成胶速度,还是在凝胶强度上都明显优于单纯的自由基聚合交联和光偶合交联构筑的水凝胶性能。从图1中看出,该溶液在约2s时开始成胶,直至10s左右完全成胶,且完全成胶时的模量可以达到3500-10000pa。此外,凝胶的强度与凝胶溶液的质量浓度成正比,凝胶的质量浓度越大,所成凝胶的强度越大。其它不同材料组成的水凝胶体系的凝胶点和凝胶强度也有所不同,具体数据如表2所示。
[0940]
表2
[0941]
[0942]
[0943]
[0944][0945]
注:nb0为文献报道的用于构筑水凝胶的邻硝基苄基类光扳机(yunlong yang;jieyuanzhang;zhenzhen liu;qiuning lin;xiaolin liu;chunyan bao;yang wang;linyong zhu.adv. mater.2016,28,2724.)。ha-nb0即为nb0标记的透明质酸高分子衍生物。nb为本发明组分a-1中的邻硝基苄基类光扳机;cnb为本发明组分a-88中的环状邻硝基苄基
类光扳机;cnb-ma为本发明组分a-144中的既含环状邻硝基苄基类光扳机又含双键官能团。其中,ha-nb即为组分a-1;ha-cnb即为组分a-88;ha-cnb-ma即为组分a-144。
[0946]
实施例一百六十八:光交联水凝胶粘附力测试
[0947]
取新鲜猪肠衣若干,将其裁剪成3.5cm
×
2.5cm大小的肠衣片。然后利用502胶水将其固定在6.5cm
×
2.5cm大小的钢化玻璃片上。取上述钢化玻璃片,在其中一片粘结肠衣面上涂抹150μl的一定组分的水凝胶前体溶液。然后,将另一片玻璃片置于此片玻璃片上方,使上下两片粘附肠衣的位置完全相对。此时,擦去多余的被挤出的水凝胶前体溶液。然后利用395nm led光源(20mw/cm2)对肠衣部位进行光照5min,使水凝胶前体溶液在两片肠衣之间原位成胶。成胶完全后,将玻璃片的一端垂直固定,另一端通过细绳连接上能够盛水的容器。随后不断向容器中加入定量水,直到两片玻璃片断开为止。其后,记录下此时水和容器的质量,将其转化成重力也就是玻璃片断裂时的拉力f,利用以下公式计算水凝胶的组织黏附力:
[0948]
水凝胶组织黏附力=f/a
[0949]
其中a为肠衣的粘接面积,测试装置示意图如图2所示。其它不同材料组成的水凝胶体系的组织粘附力也有所不同,具体数据如表3所示。
[0950]
表3
[0951]
[0952]
[0953]
[0954][0955]
实施例一百六十九:光交联水凝胶力学性能测试
[0956]
力学性能测试(包括拉伸测试和压缩测试)采用gt-tcs-2000拉力机,拉伸测试样品为长20mm,宽3mm,厚2mm的哑铃型试样,测试速度为5mm/min,压缩测试样品为直径10mm,高3mm的圆柱形试样,测试速度为1mm/min,以实施例一制备的组分 a-1(即为ha-nb)、实施例一百零七制备的组分a-107(即为hama)、组分c-4(即为明胶)、实施例一百五十五制备的组分b-2(即为lap);实施例八十八制备的组分a-88 (即为ha-cnb)、实施例一百四十四制备的组分a-144(即为ha-cnb-ma)、实施例一百五十五制备的组分b-2(即为lap)为例,测试该水凝胶的拉伸性能和压缩性能。从图 3中看出,水凝胶(ha-nb/hama/gelatin/lap)能够被压缩到75%左右,压缩强度为 2mpa左右;水凝胶(ha-cnb/ha-cnb-ma/lap)能够被压缩到88%左右,压缩强度为 1mpa左右。其它不同材料组成的水凝胶体系的力学性能也有所不同,具体数据如表4所示。
[0957]
表4
[0958]
[0959]
[0960]
[0961][0962]
实施例一百七十:光交联水凝胶生物相容性测试
[0963]
在本实验中,以实施例一制备的组分a-1(即为ha-nb)、实施例一百零七制备的组分a-107(即为hama)、组分c-4(即为明胶)、实施例一百五十五制备的组分b-2(即为lap);实施例八十八制备的组分a-88(即为ha-cnb)、实施例一百四十四制备的组分a-144(即为ha-cnb-ma)、实施例一百五十五制备的组分b-2(即为lap)为例,通过cck-8试剂盒进行评价。首先,在96孔板中种植成纤维细胞hdfs,细胞密度为5
×
103细胞/孔,然后加入培养基,在37℃/5%co2条件下培养24h。将各组测试样品溶解于细胞培养液中,加入到培养有细胞的孔
板中,继续培养24h,然后将孔中的细胞液吸出,向每个孔中加入100μl的培养基和10μl的cck-8溶液,继续孵育细胞2h。最后,用酶标仪检测每个孔中450nm的吸光度。细胞存活率计算如下:
[0964]
cell viability(%)=(实验组吸光度的平均值/控制组吸光度的平均值)
×
100%
[0965]
从图4中看出,该类光交联水凝胶具有较好的生物相容性。
[0966]
体内免疫炎症反应测试中,以实施例一制备的组分a-1(即为ha-nb)、实施例一百零七制备的组分a-107(即为hama)、组分c-4(即为明胶)、实施例一百五十五制备的组分b-2(即为lap);实施例八十八制备的组分a-88(即为ha-cnb)、实施例一百四十四制备的组分a-144(即为ha-cnb-ma)、实施例一百五十五制备的组分b-2(即为 lap)为例,将水凝胶植入兔子皮下,选取不同时间点分别通过组织切片染色分析该类水凝胶对机体产生的炎症反应。
[0967]
其它不同材料组成的水凝胶体系的生物相容性也有所不同,具体数据如表5所示。
[0968]
表5
[0969]
[0970][0971]
以上不同组分的水凝胶材料中组分a与组分b关系均为2%wt:0.2%wt。组分a与组分b与组分c关系均为2%wt:0.2%wt:2%wt。
[0972]
实施例一百七十一:光交联水凝胶应用于创面封闭-皮肤修复
[0973]
实验中,在sd大鼠背部皮肤构造直径1.8cm的皮肤完全缺损伤口。然后将400μl 水凝胶前体溶液(2%组分a-1/1%组分a-107/6%组分c-4/0.2%组分b-2)填充到伤口部位。由于该溶液具有良好的流动性,伤口可以被水凝胶前体溶液充分填充和渗透。然后,在395nm led光源照射下,在皮肤缺损处原位制备了水凝胶,实现了对创面的封闭 (如图5所示)。接下来,对比了原位成型的水凝胶,预先成型的水凝胶和仅用生理盐水清洗处理的sd
大鼠背部皮肤伤口在7天内的修复效果。原位成型的水凝胶伤口修复速率要明显快于其他两组,7d时伤口收缩的面积最大,起到了良好的修复效果。而预先成型的水凝胶材料难以充分的填充伤口部位;另外,同组织间不具有共价连接的无缝界面,缺乏良好的组织整合性。新生细胞和组织难以快速的进入到水凝胶材料中,使其充分发挥支架材料的作用。因此,预先成型的水凝胶修复速率和效果要差于原位成型的水凝胶。没有水凝胶填充的伤口修复速率最慢,说明了该光交联水凝胶作为细胞支架材料对伤口修复具有促进作用。
[0974]
其他不同材料组成的水凝胶体系(组分a:组分a-1~组分a-154;组分b:组分b-1~组分b-3;组分c-1~c-21)属于光交联水凝胶,同样可以应用于创面封闭-皮肤修复。
[0975]
实施例一百七十二:光交联水凝胶应用于创面封闭-术后防黏连
[0976]
实验中,采用sd大鼠构建腹壁-盲肠刮擦的腹腔黏连模型。由于盲肠是腹腔内最粗、通路最多,血管分布最丰富的肠段,当其对应的腹壁同时发生损伤并且不采取措施的前提下发生腹腔黏连的概率极大,构造的黏连模型稳定。在手术过程中,水凝胶前体溶液(2%组分a-1/1%组分a-107/6%组分c-4/0.2%组分b-2)可以充分的覆盖盲肠和腹壁的伤口,并且在垂直的组织面上有充足的驻留时间至其光照成胶。在给予30s光照后,得到的水凝胶固定在了创伤部位,利用外科手术刀片对其施加一定的力的情况下并不能将该水凝胶从创伤部位剥离。上述从水凝胶前体溶液的给予到完全成胶的过程在1min内可以结束(如图6所示)。手术后,在无菌的环境下饲养上述sd大鼠14天。14d后,再次打开 sd大鼠的腹腔,对其腹腔黏连情况进行了记录。在水凝胶处理的实验组的10只大鼠中,有8只大鼠14d后没有出现任何的肠-腹壁,肠-肠之间的黏连;1只大鼠出现了中等程度的腹壁与盲肠之间的黏连;1只大鼠出现了肠与肠之间一点薄层黏连。另外,上述9只没有发生肠-腹壁黏连的sd大鼠中看不见任何的水凝胶残留,腹壁上的伤口完全愈合。对照组的10只大鼠都发生了严重的腹壁和盲肠的黏连。接下来对实验组和对照组手术中伤口部位的组织切片通过h&e染色的方式进行了组织学分析。实验组中的sd大鼠14d后盲肠和腹壁的损伤基本完全恢复,表层已经再上皮化。而对照组中的sd大鼠14d后盲肠的平滑肌同腹壁的肌肉组织完全融合,成纤维细胞和炎症细胞在黏连处沉积。
[0977]
其他不同材料组成的水凝胶体系(组分a:组分a-1~组分a-154;组分b:组分b-1~组分b-3;组分c-1~c-21)属于光交联水凝胶,同样可以应用于创面封闭-术后防黏连。
[0978]
实施例一百七十三:光交联水凝胶应用于创面封闭-口腔溃疡
[0979]
实验中,在sd大鼠口腔构造直径1.0cm的口腔溃疡缺损伤口。然后将200μl水凝胶前体溶液(2%组分a-1/1%组分a-107/6%组分c-4/0.2%组分b-2)填充到伤口部位。由于该溶液具有良好的流动性,伤口可以被水凝胶前体溶液充分填充和渗透。然后,在395nm led光源照射下,在口腔缺损处原位制备了水凝胶,实现了对口腔创面的封闭。接下来,对比了原位成型的水凝胶,预先成型的水凝胶和仅用生理盐水清洗处理的sd大鼠口腔伤口在7天内的修复效果。原位成型的水凝胶伤口修复速率要明显快于其他两组, 7d时伤口收缩的面积最大,起到了良好的修复效果。而预先成型的水凝胶材料难以充分的填充伤口部位;另外,同组织间不具有共价连接的无缝界面,缺乏良好的组织整合性。新生细胞和组织难以快速的进入到水凝胶材料中,使其充分发挥支架材料的作用。因此,预先成型的水凝胶修复速率和效果要差于原位成型的水凝胶。没有水凝胶填充的伤口修复速率最慢,说明了该光交联水凝胶作为细胞支架材料对口腔溃疡修复具有促进作用。
[0980]
其他不同材料组成的水凝胶体系(组分a:组分a-1~组分a-154;组分b:组分b-1~组分b-3;组分c-1~c-21)属于光交联水凝胶,同样可以应用于创面封闭-口腔溃疡。
[0981]
实施例一百七十四:光交联水凝胶应用于组织渗液封堵-肠漏封堵
[0982]
采用新西兰雄性大白兔,分为两组进行盲肠渗漏封堵实验:a:水凝胶处理(2%组分a-1/1%组分a-107/6%组分c-4/0.2%组分b-2)组;b:不做处理的对照组。实验中,在兔子盲肠处制造渗漏的模型,然后将水凝胶前体溶液涂抹到伤口处,待充分渗透后光照原位成胶,成胶后水凝胶能牢固的黏附在缺损处,不需要额外的固定。在手术4周后,通过静脉注射空气的方法处死实验中的兔子,并提取盲肠对实验修复效果进行评价。结果显示,使用水凝胶封堵的盲肠没有发生渗漏的情况,而没用水凝胶处理的盲肠发生了严重的渗漏。经过几周的修复,原来盲肠有缺损的部位经水凝胶处理过后得到了明显的修复,因此,该水凝胶不仅能够有效封堵渗漏,还有利于术后受损组织的修复。
[0983]
其他不同材料组成的水凝胶体系(组分a:组分a-1~组分a-154;组分b:组分b-1~组分b-3;组分c-1~c-21)属于光交联水凝胶,同样可以应用于组织渗液封堵-肠漏封堵。
[0984]
实施例一百七十五:光交联水凝胶应用于组织渗液封堵-手术缝合
[0985]
采用新西兰雄性大白兔,分为三组进行手术缝合实验:a:水凝胶处理(2%组分a-1/1%组分a-107/6%组分c-4/0.2%组分b-2)组;b:手术缝合线处理组;c:不做处理的对照组。实验中,在兔子腹部制造伤口缝合的模型,a组是将水凝胶前体溶液涂抹到伤口处,待充分渗透后光照原位成胶,实现对伤口的封堵,由于该水凝胶优异的组织粘附力,能够起到组织缝合的效果;b组是用常规的手术缝合线处理伤口;c组是对伤口不做处理。在手术2周后,通过静脉注射空气的方法处死实验中的兔子,并取样对实验修复效果进行评价。结果显示,使用水凝胶处理的伤口有较好的缝合效果,基本与手术缝合线组的效果差不多,而不做处理的伤口不能有效连接到一起。经过4周的修复,原来伤口缺损部位经水凝胶处理过后组织能够连接到一起,并且得到了明显的修复,因此,该水凝胶不仅能够有效缝合伤口,还有利于术后受损组织的修复。
[0986]
其他不同材料组成的水凝胶体系(组分a:组分a-1~组分a-154;组分b:组分b-1~组分b-3;组分c-1~c-21)属于光交联水凝胶,同样可以应用于组织渗液封堵-手术缝合。
[0987]
实施例一百七十六:光交联水凝胶应用于止血材料-肝脏止血
[0988]
采用sd大鼠,对水凝胶的止血效果进行评价,分为三组进行肝脏止血实验:a:明胶海绵组;b:水凝胶处理(2%组分a-1/1%组分a-107/6%组分c-4/0.2%组分b-2) 组;c阳性对照组。实验大鼠通过水合氯醛(4%水溶液)腹腔注射进行麻醉,注射计量为0.9ml/100g,深度麻醉后,用剃毛器将大鼠前胸部位毛剃光,碘酒消毒。然后沿着胸腔中线切开大约4cm长切口,打开胸腔,暴露肝脏部位。在肝脏左叶做一约2cm切口。 a组用明胶海绵进行止血;b组在切口处加水凝胶前体溶液覆盖切面,395nm led光照2 min成胶止血;c组不做任何处理,让肝脏切口渗血自然凝固,用纱布吸去渗血,通过减重法记录出血量,和出血时间(如图7所示)。实验结束后,a组将粘附在切面的明胶海绵一并留在大鼠体内进行缝合。b组水凝胶在切口原位交联并将切面伤口隔离,将肝脏放回胸腔,缝合。c组不做处理直接缝合。14d后,观察sd大鼠肝脏恢复情况,通过腹腔注射过量麻醉剂水合氯醛(4%水溶液,2.7ml/100g)处死大鼠,沿胸腔中线打开胸腔,观察三组大鼠肝脏恢复情况,并拍照记录。同时对肝脏损伤部位组织取样,标本用4%福尔马林溶液固定2d,脱水处理后,石蜡包埋,在用切片机进行组
织切片操作,样片厚度 5μm。最后对标本进行h&e染色,用光学显微镜拍照观察记录。实验结果显示,b组肝脏恢复良好,水凝胶降解完全,未发生粘连,肝脏切口长出新生肝脏组织。a组大鼠体内明胶海绵仍未降解,并且大鼠普遍器脏与网膜粘连严重。c组普遍存在肝脏与网膜粘连的情况。h&e染色显示实验组肝脏表面光滑圆润,有丰富的血管分布,肝脏界面清晰。而发生粘连的肝脏经h&e染色发现肝脏界面凹凸不平,肝脏与网膜组织粘连在一起,界面处有沉积的炎症细胞。
[0989]
其他不同材料组成的水凝胶体系(组分a:组分a-1~组分a-154;组分b:组分b-1~组分b-3;组分c-1~c-21)属于光交联水凝胶,同样可以应用于止血材料-肝脏止血。
[0990]
实施例一百七十七:光交联水凝胶应用于止血材料-骨断面止血
[0991]
采用新西兰雄性大白兔,分为三组进行骨断面止血实验:a:水凝胶处理(2%组分 a-1/1%组分a-107/6%组分c-4/0.2%组分b-2)组;b:骨蜡处理组;c:不做处理的对照组。实验中,在兔子股骨制造骨断面出血模型,a组是将水凝胶前体溶液涂抹到伤口处,待充分渗透后光照原位成胶,实现对骨断面出血的有效封堵,由于该水凝胶优异的组织粘附力及光固化速度,能够起到及时、有效的止血效果;b组是用常规的骨蜡处理出血伤口; c组是对出血伤口不做处理。在手术8周后,通过静脉注射空气的方法处死实验中的兔子,并取样对实验修复效果进行评价。结果显示,使用水凝胶处理的伤口有较好的止血效果,基本与骨蜡组的效果差不多,而不做处理的伤口会有持续的出血状况。经过2周的修复,原来伤口出血部位经水凝胶处理过后组织得到了明显的修复,而经骨蜡处理的伤口没有得到修复,主要是由于骨蜡在体内不降解,因此,该水凝胶不仅能够有效实现骨断面止血,还有利于术后受损组织的修复。
[0992]
其他不同材料组成的水凝胶体系(组分a:组分a-1~组分a-154;组分b:组分b-1~组分b-3;组分c-1~c-21)属于光交联水凝胶,同样可以应用于止血材料-骨断面止血。
[0993]
实施例一百七十八:光交联水凝胶应用于止血材料-动脉止血
[0994]
采用新西兰雄性大白兔,分为三组进行动脉止血实验:a:水凝胶处理(2%组分a-1/1%组分a-107/6%组分c-4/0.2%组分b-2)组;b:止血钳处理组;c:不做处理的对照组。实验中,在兔子股动脉制造出血模型,a组是将水凝胶前体溶液涂抹到伤口处,待充分渗透后光照原位成胶,实现对股动脉出血的有效封堵,由于该水凝胶优异的组织粘附力及光固化速度,能够起到及时、有效的止血效果;b组是用常规的止血钳处理出血伤口;c组是对出血伤口不做处理。在手术2周后,通过静脉注射空气的方法处死实验中的兔子,并取样对实验修复效果进行评价。结果显示,使用水凝胶处理的伤口有较好的止血效果,基本与止血钳的效果差不多,而不做处理的伤口会有持续的出血状况。经过2周的修复,原来伤口出血部位经水凝胶处理后组织得到了明显的修复,因此,该水凝胶不仅能够有效实现股动脉止血,还有利于术后受损组织的修复。
[0995]
其他不同材料组成的水凝胶体系(组分a:组分a-1~组分a-154;组分b:组分b-1~组分b-3;组分c-1~c-21)属于光交联水凝胶,同样可以应用于止血材料-动脉止血。
[0996]
实施例一百七十九:光交联水凝胶应用于止血材料-心脏止血
[0997]
采用新西兰雄性大白兔,分为三组进行心脏止血实验:a:水凝胶处理(2%组分a-1/1%组分a-107/6%组分c-4/0.2%组分b-2)组;b:明胶海绵处理组;c:不做处理的对照组。实验中,在兔子心脏制造出血模型,a组是将水凝胶前体溶液涂抹到伤口处,待充分渗透
后光照原位成胶,实现对心脏出血的有效封堵,由于该水凝胶优异的组织粘附力及光固化速度,能够起到及时、有效的止血效果;b组是用常规的明胶海绵处理出血伤口;c 组是对出血伤口不做处理。在手术2周后,通过静脉注射空气的方法处死实验中的兔子,并取样对实验修复效果进行评价。结果显示,使用水凝胶处理的伤口有较好的止血效果,要比明胶海绵的止血效果好,而不做处理的伤口会有持续的出血状况。经过2周的修复,原来伤口出血部位经水凝胶处理后组织得到了明显的修复,并且修复效果比明胶海绵好,因此,该水凝胶不仅能够有效实现心脏止血,还有利于术后受损组织的修复。
[0998]
其他不同材料组成的水凝胶体系(组分a:组分a-1~组分a-154;组分b:组分b-1~组分b-3;组分c-1~c-21)属于光交联水凝胶,同样可以应用于止血材料-心脏止血。
[0999]
实施例一百八十:光交联水凝胶应用于组织工程支架材料-软骨修复
[1000]
采用新西兰雄性大白兔,分为三组进行关节软骨的修复实验:a:包裹有软骨细胞的水凝胶(2%组分a-1/1%组分a-107/6%组分c-4/0.2%组分b-2)组,即gel+软骨细胞组;b:单纯的水凝胶(2%组分a-1/1%组分a-107/6%组分c-4/0.2%组分b-2)组,即gel组;c:不做处理的对照组,即control组。在实验中,该水凝胶前体溶液可以充分的渗透并且填充兔子关节软骨的缺损处,光照成胶后牢固的黏附在缺损处,不需要额外的固定。在手术12周后,通过静脉注射空气的方法处死实验中的兔子,并提取损伤关节对实验修复效果进行评价。兔子关节软骨损伤处大体观照片结果显示,12周后gel+软骨细胞组在关节缺损处长出了光滑的新生软骨组织,同时和旧的软骨组织进行了良好的整合;在gel组中软骨也进行了一定的修复,但是还可以看出手术时软骨创伤的轮廓;而在 control组中,软骨组织基本没有修复的情况,损伤处还是明显的空洞。接下来,我们进一步利用h&e染色的方法评价了上述各组软骨的修复情况。h&e染色结果显示,gel+ 软骨细胞组和gel组都有新生的组织生成并且同旧的软骨组织整合良好;但是gel+软骨细胞组的新生组织的厚度要好于gel组,并且表面平整;而在control组中难以找到明显新生组织的迹象。另外,采用番红-o和免疫组化染色的方法对新生软骨的成分进行了分析。在gel+软骨细胞组和gel组中,新生的软骨组织都表现出了番红-o染色活性,证明该新生的软骨组织内含有正常软骨的糖蛋白成分。同时,gel+软骨细胞组和gel组的新生软骨组织都表现出ii型胶原的染色活性,证明该软骨组织中含有大量的ii型胶原。上述番红-o和免疫组化染色结果证明利用新型光交联水凝胶材料进行软骨修复时,新生的软骨组织是透明软骨。
[1001]
其他不同材料组成的水凝胶体系(组分a:组分a-1~组分a-154;组分b:组分b-1~组分b-3;组分c-1~c-21)属于光交联水凝胶,同样可以应用于组织工程支架材料-软骨修复。
[1002]
实施例一百八十一:光交联水凝胶应用于组织工程支架材料-骨修复
[1003]
采用sd大鼠,进行颅骨修复实验,并将上述sd大鼠随机分成3组:a:水凝胶(2%组分a-1/1%组分a-107/6%组分c-4/0.2%组分b-2)+羟基磷灰石的实验组;b:水凝胶处理(2%组分a-1/1%组分a-107/6%组分c-4/0.2%组分b-2)组;c:不用材料处理的对照组。实验中,用4%的水合氯醛溶液(0.9ml每克体重)对其进行腹腔麻醉,碘酒消毒。然后,利用外科手术刀片打开大鼠颅骨处头皮。利用牙环钻在老鼠颅骨左右处对称制造直径5mm的完全颅骨缺损模型。在实验组中,取200μl的水凝胶前体溶液填充到sd大鼠颅骨缺损处,使其充分向伤口边缘渗透;用395nm led光源(20mw/cm2) 光照30s使其完全成胶;最后用缝合线缝
合老鼠的头皮。在对照组中,制造好sd大鼠颅骨缺损模型后,直接缝合头皮,不做其他任何处理。上述sd大鼠在无菌,37℃的环境中饲养8周的时间。然后,利用micro-ct扫描成像的方式对各组中sd大鼠颅骨的修复情况进行了评价。结果显示,在没有进行任何处理的对照组中,sd大鼠的颅骨缺损基本没有进行任何的修复,而用水凝胶填充的颅骨缺损处边缘有新生的成骨形成,但是新生骨组织的量较少,大部分缺损处并没有得到良好的修复,而用水凝胶+羟基磷灰石填充的颅骨缺损处基本得到了修复,大量的新生骨组织在缺损处形成。接着利用van gieson染色法对颅骨的组织切片进行了组织学染色分析。结果显示,水凝胶+羟基磷灰石处理的sd 大鼠的颅骨缺损处都长出了完整的新生骨组织,而只用水凝胶处理的颅骨缺损处只有少量新生骨组织生成,大部分缺损处的骨组织依旧是缺损状态,在对照组中,几乎没有新生的骨组织生成。该组织染色结果进一步证实了包裹有羟基磷灰石的水凝胶对骨缺损有良好的修复效果。
[1004]
其他不同材料组成的水凝胶体系(组分a:组分a-1~组分a-154;组分b:组分b-1~组分b-3;组分c-1~c-21)属于光交联水凝胶,同样可以应用于组织工程支架材料-骨修复。
[1005]
实施例一百八十二:光交联水凝胶应用于组织工程支架材料-骨/软骨复合缺损修复
[1006]
采用猪作为动物模型,软骨相材料为水凝胶+软骨细胞,骨相材料为水凝胶+羟基磷灰石+bmscs,分为三组进行关节骨/软骨复合缺损的修复实验:a:分别包裹有软骨细胞和bmscs的水凝胶(2%组分a-1/1%组分a-107/6%组分c-4/0.2%组分b-2)组,即gel+细胞组;b:单纯的水凝胶(2%组分a-1/1%组分a-107/6%组分c-4/0.2%组分 b-2)组,即gel组。在实验中,先将骨相材料填充至骨相缺损处,并使凝胶前体溶液充分渗透,光照成胶后水凝胶牢固的粘附在骨缺损处,然后将软骨相材料填充至软骨相缺损处,光照成胶后水凝胶牢固的黏附在软骨缺损处(如图8所示)。在手术6个月后,处死实验的猪,并提取损伤关节对实验修复效果进行评价。gel+细胞组在关节缺损处长出了光滑的新生软骨组织和骨组织,并且和旧的软骨/骨组织进行了良好的整合,同时,软骨组织和骨组织也进行了良好的整合;在gel组中骨/软骨组织基本没有修复的情况,损伤处还是明显的空洞。接下来,进一步利用h&e染色的方法评价了上述各组软骨的修复情况。h&e染色结果显示,gel+细胞组有新生的组织生成并且同旧的软骨组织整合良好;但是在gel组中难以找到明显新生组织的迹象。另外,采用番红-o和免疫组化染色的方法对新生软骨的成分进行了分析。在gel+细胞组中,新生的软骨组织都表现出了番红-o 染色活性,证明该新生的软骨组织内含有正常软骨的糖蛋白成分。同时,gel+细胞组的新生软骨组织都表现出ii型胶原的染色活性,证明该软骨组织中含有大量的ii型胶原。上述番红-o和免疫组化染色结果证明利用新型光交联水凝胶材料进行软骨修复时,新生的软骨组织是透明软骨。接着利用van gieson染色法对骨的组织切片进行了组织学染色分析。结果显示,gel+细胞组处理的骨缺损处长出了完整的新生骨组织,而在gel组中骨缺损处只有少量新生骨组织生成,大部分缺损处的骨组织依旧是缺损状态。该组织染色结果进一步证实了加细胞的水凝胶对骨缺损有良好的修复效果。
[1007]
其他不同材料组成的水凝胶体系(组分a:组分a-1~组分a-154;组分b:组分b-1~组分b-3;组分c-1~c-21)属于光交联水凝胶,同样可以应用于组织工程支架材料-骨/ 软骨复合缺损修复。
[1008]
实施例一百八十三:光交联水凝胶应用于3d打印(fdm)的生物墨水
[1009]
3d打印技术是近些年来迅速发展的一种三维成型技术,已被广泛应用,目前3d打印技术包括熔融沉积式(fdm)、光固化成型(sla)、激光烧结式(sls)、连续液面制造式(clip)等。但是适用于带细胞打印的方式目前主要是fdm的方式,带细胞打印的材料主要是水凝胶材料,因此,发展3d打印的生物墨水-可打印的水凝胶材料以及提高水凝胶材料打印的分辨率是该领域研究的基本问题。以实施例一制备的组分a-1(即为 ha-nb)、实施例一百零七制备的组分a-107(即为hama)、组分c-4(即为明胶)、实施例一百五十五制备的组分b-2(即为lap);实施例八十八制备的组分a-88(即为 ha-cnb)、实施例一百四十四制备的组分a-144(即为ha-cnb-ma)、实施例一百五十五制备的组分b-2(即为lap)为例,将一定质量浓度的水凝胶前体溶液均匀混合细胞后,装入低温打印桶中,控制打印温度在25℃左右,通过温度来调整生物墨水的粘稠度,以获得最佳的打印状态,然后确定合适的打印压力和打印速度,进行不同结构的生物打印,打印完成后通过光照交联水凝胶(或是边打印边光照),从而获得带细胞且带结构的水凝胶,进行3d细胞培养(如图9所示)。
[1010]
其他不同材料组成的水凝胶体系(组分a:组分a-1~组分a-154;组分b:组分b-1~组分b-3;组分c-1~c-21)属于光交联水凝胶,同样可以应用于3d打印(fdm)的生物墨水。
[1011]
实施例一百八十四:光交联水凝胶应用于3d打印(dlp)的生物墨水
[1012]
dlp(数字光处理)3d打印技术是近来年发展起来的一种新型的光固化打印方式,相比于sla(立体光固化成型)式的打印机,dlp以其打印速度快、分辨率高而具有大多数打印方式所不可比拟的优势,目前在牙科模型、珠宝设计等领域已经具备一定的应用前景。但是,当前市场上使用的打印墨水仅限于光固化树脂,而水凝胶作为一种新兴的生物墨水还没有得到广泛的关注,主要是由于没有适合dlp打印的水凝胶材料,而本发明提出的复合型光交联水凝胶材料以其快的光固化速度,优异的机械性能非常适合于3d打印,而且具有更高的打印精度。以实施例一制备的组分a-1(即为ha-nb)、实施例一百零七制备的组分a-107(即为hama)、组分c-4(即为明胶)、实施例一百五十五制备的组分b-2(即为lap);实施例八十八制备的组分a-88(即为ha-cnb)、实施例一百四十四制备的组分a-144(即为ha-cnb-ma)、实施例一百五十五制备的组分b-2(即为 lap)为例,将一定质量浓度的水凝胶前体溶液均匀混合细胞后,装入液体槽中,通过控制光源的强弱、曝光时间等参数来调整生物墨水的打印情况,以获得最佳的打印状态,从而得到了既带细胞又带结构的水凝胶,可以进行3d细胞培养的研究。
[1013]
其他不同材料组成的水凝胶体系(组分a:组分a-1~组分a-154;组分b:组分b-1~组分b-3;组分c-1~c-21)属于光交联水凝胶,同样可以应用于3d打印(dlp)的生物墨水。
[1014]
实施例一百八十五:光交联水凝胶应用于药物的包裹与释放
[1015]
水凝胶是一种能够在水中溶胀但不溶解的交联高分子网络,由于水凝胶大部分由水组成,因此具有非常好的生物相容性,特别适用于药物和生物活性大分子的载体。包裹于水凝胶材料中的药物或生物活性大分子通过分子的扩散作用和材料的降解作用实现药物持续释放的效果。以药物包裹与释放为例具体介绍如下:以实施例一制备的组分a-1(即为 ha-nb)、实施例一百零七制备的组分a-107(即为hama)、组分c-4(即为明胶)、实施例一百五十五制备的组分b-2(即为lap);实施例八十八制备的组分a-88(即为 ha-cnb)、实施例一百四十四制备的组分a-144(即为ha-cnb-ma)、实施例一百五十五制备的组分b-2(即为lap)为例,将其溶于生理盐水中,配成一定质量浓度的水凝胶前体溶液,加入一定量的药物分子,
取200μl上述溶液置于圆形模具中光照成水凝胶,接着放入24孔细胞培养板中,加入一定量的生理盐水进行药物释放实验,通过紫外测试分析溶液中药物的释放量,以此来评价该材料对药物的释放效果。
[1016]
其他不同材料组成的水凝胶体系(组分a:组分a-1~组分a-154;组分b:组分b-1~组分b-3;组分c-1~c-21)属于光交联水凝胶,同样可以应用于药物的包裹与释放。
[1017]
上述的对实施例的描述是为便于该技术领域的普通技术人员能理解和使用发明。熟悉本领域技术的人员显然可以容易地对这些实施例做出各种修改,并把在此说明的一般原理应用到其他实施例中而不必经过创造性的劳动。因此,本发明不限于上述实施例,本领域技术人员根据本发明的揭示,不脱离本发明范畴所做出的改进和修改都应该在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1