一种熊蜂生假丝酵母强组成型启动子及其应用的制作方法
[0001]
本发明涉及一种熊蜂生假丝酵母强组成型启动子及其应用,属于分析化学技术领域。
背景技术:
[0002]
熊蜂生假丝酵母(starmerella bombicola)是一种非致病性的单倍体假丝酵母,因其生产的槐糖脂具有良好的降低表面张力、起泡性、提高溶解性等特性而被广泛的应用到农业、医药及食品行业等领域,并且因其良好的环境友好性,在环境保护方面有着十分广泛的应用前景。目前,国内外相关的研究已经基本阐明了熊蜂生假丝酵母生物合成槐糖脂的代谢途径,并且利用代谢工程的方法提高槐糖脂的产量。现有的技术以主要是利用过表达槐糖脂合成的关键基因提高其产量,而启动子在基因表达过程中发挥着十分重要的作用,控制着基因转录水平的高低。
[0003]
现如今,随着合成生物学的发展,人们也越来越关注启动子等合成生物学元件领域的相关研究,而强启动子在异源基因的高效表达中有着非常关键的作用,并且组成型启动子的调控不受外界条件的影响,能够使所启动基因持续表达,因此更具有良好的应用前景。熊蜂生假丝酵母中,关于启动子的研究较少,因而在很大程度上限制了其启动子应用的前景,需进一步分析及鉴定熊蜂生假丝酵母启动子并获得强组成型启动子,为其在外源基因高效表达、基因工程及合成生物学等领域奠定基础。
技术实现要素:
[0004]
为解决上述问题,本发明在熊蜂生假丝酵母中,以报告基因对其启动子进行预测并克隆、筛选及应用评价,并获得了一个强组成型启动子。
[0005]
本发明的第一个目的是提供一种熊蜂生假丝酵母强组成型启动子,所述启动子的核苷酸序列如seq id no.1所示。
[0006]
本发明的第二个目的是提供一种包含所述启动子的表达载体。
[0007]
进一步地,所述的表达载体为适用于熊蜂生假丝酵母组成型表达的载体。
[0008]
进一步地,所述的表达载体是以整合型质粒pphp、整合型质粒pshs或整合型质粒paha为出发载体。
[0009]
进一步地,所述的整合型质粒pphp的核苷酸序列如seq id no.2所示。
[0010]
本发明的第三个目的是提供一种包含所述启动子的熊蜂生假丝酵母。
[0011]
本发明的第四个目的是提供所述启动子在熊蜂生假丝酵母遗传改造中的应用。
[0012]
进一步地,所述的应用是采用所述启动子启动组成型基因的表达。
[0013]
进一步地,所述的应用具体包括如下步骤:
[0014]
s1、将所述启动子、待表达基因及终止子进行融合pcr得到基因片段;
[0015]
s2、将s1步骤得到的基因片段连接到整合型质粒上,得到整合型表达载体;
[0016]
s3、将s2步骤得到的整合型表达载体转入熊蜂生假丝酵母宿主菌中,得到待表达
基因增强表达的熊蜂生假丝酵母重组菌。
[0017]
进一步地,所述的终止子的核苷酸序列如seq id no.4所示。
[0018]
本发明的有益效果:
[0019]
本发明以熊蜂生假丝酵母为研究对象,预测、克隆、筛选、评价强组成型启动子。以增强型绿色荧光蛋白为报告基因,通过上述所选启动子驱动其表达,分析绿色荧光强度差异及转录水平差异,最终获得了一个具有强转录活性的组成型启动子px。该启动子与已知强组成型pgpd启动子相比,在以葡萄糖或油酸为唯一碳源及不同培养时期均具有最高强度的转录活性。该实验在熊蜂生假丝酵母中首次以增强型绿色荧光蛋白为报告基因,通过转录组水平分析得到一段具有转录活性强的dna序列,具有良好的应用前景,丰富了合成生物学元件,同时也为该菌种在其基因工程改造及外源基因表达奠定了理论基础。
附图说明
[0020]
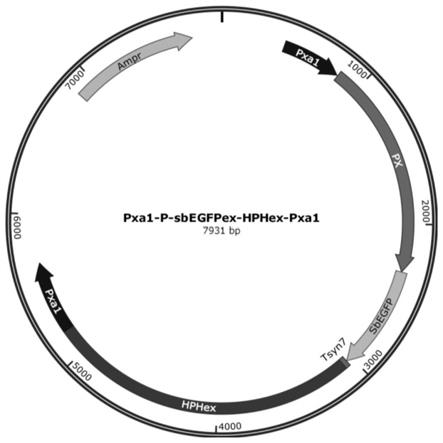
图1为所构建的整合型启动子绿色荧光报告载体示意图;
[0021]
图2为重组菌株在ypd培养基中培养绿色荧光观察图;
[0022]
图3为重组菌株在葡萄糖为唯一碳源培养基中培养8小时所测绿色荧光强度数据及转录水平数据。
[0023]
图4为重组菌株在油酸为唯一碳源培养培养基中培养8小时所测绿色荧光强度数据及转录水平数据。
[0024]
图5为重组菌株在分别以葡萄糖和油酸为唯一碳源培养80小时所测得绿色荧光蛋白强度数据图。
具体实施方式
[0025]
下面结合具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
[0026]
下述示例所用的培养基配方:(1)ypd培养基(g/l):酵母粉10,蛋白胨20,葡萄糖20。油酸培养基(g/l):酵母粉10,蛋白胨20,菜籽油60。(2)lb培养基(g/l):酵母粉5,蛋白胨10,氯化钠10。(3)潮霉素抗性培养基(μg/ml):将潮霉素母液加入到冷却至46℃左右的ypd培养基中,终浓度为500。(4)氨苄青霉素抗性培养基(g/l):氨苄青霉素加入至lb培养基中,终浓度为0.1。
[0027]
下述示例所用试剂及仪器:一步连接试剂盒购自南京诺唯赞公司,潮霉素购自北京鼎国生物有限公司;dna聚合酶、荧光定量分析试剂盒、酵母total rna提取试剂盒及核酸内切酶均购自takara公司;micropulser电转仪购自伯乐生命医学(上海)有限公司bio-rad;激光共聚焦显微镜leica tcs sp8(德国徕卡公司);流式细胞仪bd facsariaⅲ(美国碧迪公司);实时荧光定量基因扩增仪cfx96(美国伯乐公司)。
[0028]
表1引物序列
[0029][0030]
实施例1:px强组成型启动子序列的获取
[0031]
1、取培养于ypd液体培养基中,不同培养时间(4、8、12、16、20h)的野生型熊蜂生假丝酵母培养液,利用takara rna提取试剂盒分别提取total rna,逆转录后得到5个不同时间点的cdna样本,交由苏州金唯智生物科技有限公司进行转录水平测定及测序分析。
[0032]
2、将分析获得的样本数据经测序质量分析筛选过滤后,利用软件计算所测基因的count值,通过python脚本和fpkm(fragments per kilobase million)公式计算每个样本中每个基因的fpkm值。最终得到一段转录水平较高的基因序列。
[0033]
3、将该基因序列与熊蜂生假丝酵母全基因组对比分析,最终选取该基因起始密码子上游1500bp作为为启动子功能区,通过设计特异性引物,px-f/px-r以熊蜂生假丝酵母基因组为模板,pcr扩增得到该启动子dna序列产物px,将该dna产物连接到pm-19t simple载体并进行测序,px测序结果如seqidno.1所示。
[0034]
实施例2:px表达gfp重组质粒构建
[0035]
1、通过熊蜂生假丝酵母密码子偏好性分析,将密码子优化并化学合成后的的yegfp基因(sbegfp)(seq id no.3)通过设计特异性引物其与启动子px进行融合pcr,最终得到绿色荧光蛋白表达盒px-yegfp-tsyn7(启动子与yegfp基因间插入bamhi酶切位点)。
[0036]
2、以本实验室前期所构建潮霉素抗性基因(hph)为筛选标记的整合型质粒pphp为出发载体(seq id no.2),经mlui酶切后回收,并与步骤1中的表达盒经c113一步连接酶连接转化大肠杆菌jm109,并涂布于氨苄青霉素选择平板,最终获得整合型启动子绿色荧光报告载体pphp-px-yegfp。
[0037]
3、以启动子绿色荧光报告载体pphp-px-yegfp为骨架,将该载体经内切酶mlui及bamhi共同消化,设计特异性引物pgapd-f/pgapd-r,熊蜂生假丝酵母基因组为模板,pcr扩增获得pgpd启动子产物,将二者进行一步连接,获得重组质粒并转入jm109感受态细胞中,最终获得上述整合型启动子报告载体pphp-pgpd-yegfp,如图1所示。
[0038]
实施例3:重组菌株构建
[0039]
1、分别以上述构建成功的整合型启动子绿色荧光报告载体为模板,pxa1基因同源臂上下游引物,pcr扩增得到整合表达盒pxa1-px-yegfpex-hphex-pxa1和pxa1-pgpd-yegfpex-hphex-pxa1 dna片段,纯化备用。
[0040]
2、熊蜂生假丝酵母电转化方法如下:
[0041]
1)接种熊蜂生假丝酵母菌落于5ml的ypd液体培养基中,于30℃、200r
·
min-1
摇床
中,培养15-20h,吸取1ml种子液接种于50ml的ypd液体培养基中,过夜培养至菌浓od
600
=1.0。
[0042]
2)4℃环境下,离心弃上清收集菌体,用15ml的预冷无菌水洗涤菌体2次。
[0043]
3)加入4ml 1m山梨醇(预冷)使菌体重悬,4℃下离心收集菌体。
[0044]
4)加入4ml现配(0.1m)liac溶液(3500μl水,400μl liac,100μl dtt),悬浮菌体,室温静置12~15min,离心收集菌体。
[0045]
5)加4ml山梨醇(1m)重悬并离心收集菌体;加入250μl山梨醇悬浮菌体。
[0046]
6)取50μl菌悬液、加入上述10μl待转化片段,到1.5ml ep管中混匀,冰浴5min;转移至预冷的1mm电转杯中,冰上放置3~5min,轻轻擦干电转杯,然后利用电转仪进行电击(1.5kv、200ω、5ms电容25μl),取出后立即加入1ml预冷的山梨醇吹吸再转入到1.5ml ep管中,30℃水浴锅中,静置孵育1h。
[0047]
7)立即取出菌悬液,吸取200μl涂布在潮霉素抗性ypd平板上,倒置放入30℃培养箱中,培养3d,至平板上长出单菌落。
[0048]
3、启动子筛选菌株的鉴定:
[0049]
1)挑取单菌落于5ml的ypd液体培养基中,30℃培养12-24h,至菌体浓度及od600达到1.0或以上。
[0050]
2)离心去上清液后收集菌体,加入600μl的山梨醇-na2edta溶液并使菌体悬浮,加入60μl蜗牛酶(50mg
·
ml-1),混匀,置于37℃恒温箱中,破壁4h,每隔1h翻转一次使细胞破壁完全。
[0051]
3)离心弃上清,加入500μl tris-hcl-na2edta溶液并使菌体悬浮,加入50μl 10%sds溶液,混匀,置于65℃恒温箱中,45min,每隔10min翻转一次(破坏细胞壁,裂解核酸,较高温下破坏蛋白质与dna的结合,使dna释放)。
[0052]
4)45min后菌液变澄清,加入300μl kac溶液(kac 5mol
·
l-1
),混匀,置于冰上放置1h(调节ph,促进蛋白质沉淀)。
[0053]
5)离心取上清液550μl于新的ep管,加入等体积的异丙醇,混匀,颠倒室温下放置15min。
[0054]
6)离心收集沉淀,用200μl 70%乙醇清洗沉淀两次。
[0055]
7)在37℃下烘干5min,加入30-35μl水,3μl rna消化酶(rnase a)后,置于37℃恒温箱中放置1h,作为模板进行pcr验证,最终得到重组菌株分别命名为px-1及pgpd-1。
[0056]
实施例4:
[0057]
1、重组菌株px-1及pgpd-1分别在以葡萄糖为唯一碳源条件下培养,绿色荧光强度测定及转录水平测定:
[0058]
1)重组菌株培养:将上述两种重组菌株接种至10ml ypd液体培养基30℃培养过夜后,分别转接至50ml以葡萄糖为唯一碳源的液体培养基中,30℃培养8h,od600达到1.0或以上,4℃冰箱放置2h。
[0059]
2)各收集1ml菌体离心用pbs缓冲液洗涤菌体2-3次,最终稀释菌体浓度到适宜浓度制片。
[0060]
3)使用leica tcs sp8激光共聚焦显微镜进行绿色荧光观察观察,参数设置为:wavelength:激发=488nm,发射=510nm,观察结果如图2所示。
[0061]
4)按上述方法,获得各重组菌株适宜浓度的菌悬液,使用流式细胞仪bd facsariaⅲ进行上样测定其荧光强度,检测条件为:fitc通道,激发488nm(blue laser照射),发射530/30,数据结果如图3(a)所示。
[0062]
5)取上述条件培养8小时的重组菌菌液,根据酵母total rna提取试剂盒说明书,提取total rna并逆转录,以actin激动蛋白基因作为参照基因,进行rt-qpcr进行其gfp转录水平测定分析,设计特异性引物actin-f/actin-r;q-yegfp-f/q-yegfp-r。实时荧光定量基因扩增仪cfx96,pcr条件为:95℃2min;96℃10s,60℃10s,72℃20s,80℃;96℃至80℃进行40个循环;61℃5min。转录水平数据如图3(b)所示。
[0063]
以同样的测定方法,将重组菌株px-1及pgpd-1分别在油酸为唯一碳源的培养基中培养,测定其绿色荧光强度及转录强度。测定结果如图4所示。
[0064]
2、重组菌株px-1及pgpd-1分别在以葡萄糖或油酸为唯一碳源条件下不同时间点荧光强度的测定
[0065]
按上述重组菌株在50ml ypd液体培养基中连续培养,每间隔8小时取样并使用流式细胞仪bd facsariaⅲ(美国碧迪公司)分别测定不同时间点各重组菌株荧光强度值,检测条件与上述一致,结果如图5所示,其中图5(a)是以葡萄糖为唯一碳源的检测结果,图5(b)是以油酸为唯一碳源的检测结果。
[0066]
由实施例可知,本发明以熊蜂生假丝酵母为研究对象,预测、克隆、筛选、评价强组成型启动子。以增强型绿色荧光蛋白为报告基因,通过上述所选启动子驱动其表达,分析绿色荧光强度差异及转录水平差异,最终获得了一个具有强转录活性的组成型启动子px。该启动子与已知强组成型pgpd启动子相比,在以葡萄糖或油酸为唯一碳源及不同培养时期均具有最高强度的转录活性。该实验在熊蜂生假丝酵母中首次以增强型绿色荧光蛋白为报告基因,通过转录组水平分析得到一段具有转录活性强的dna序列,具有良好的应用前景,丰富了合成生物学元件,同时也为该菌种在其基因工程改造及外源基因表达奠定了理论基础。
[0067]
以上所述实施例仅是为充分说明本发明而所举的较佳的实施例,本发明的保护范围不限于此。本技术领域的技术人员在本发明基础上所作的等同替代或变换,均在本发明的保护范围之内。本发明的保护范围以权利要求书为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1