突变Aerolysin单体在检测RNA碱基序列及RNA修饰方面的应用的制作方法
本发明涉及基因检测技术领域,涉及纳米孔检测rna修饰方法,具体为突变aerolysin单体在检测rna碱基序列及rna修饰方面的应用。
技术背景
目前开发应用范围跨度广、快速且廉价的核酸鉴别技术是必要的。现有技术检测慢、价格昂贵的主要原因是它们需要扩增技术以产生大量体积的核酸,并且需要大量的用于信号检测的专业荧光化学试剂。纳米孔作为直接的电生物传感器,可降低所需核苷酸和试剂的数量,在快速、廉价地实现核酸检测方面具有巨大的潜力。
纳米孔传感技术通过检测待测物分子与孔道的相互作用产生的特征电流阻断信号来实现单分子水平的分析。当在纳米孔上施加电压时,当存在待测物,例如核酸的情况下,会产生电流的变化。通过分析物的特征电流标志,例如阻断电流大小和持续时间来揭露分析物的特性。使用纳米孔进行核酸检测的两个基本组分是(1)控制通过孔的核酸移动和(2)在核酸聚合物通过孔移动时鉴别核苷酸。
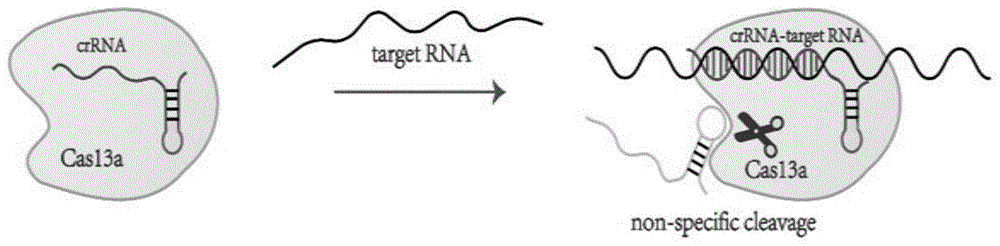
crispr是目前流行的强大的基因编辑工具,一些cas酶例如cas9,靶向dna,而另一些cas酶,例如cas13a(又称c2c2)可以与crisprrna(cr-rna)重编程,以特异性识别rna。识别其rna靶标后,激活cas13a参与附近非靶标rna的“附带”切割。该方法可以通过设计cr-rna序列,根据纳米孔对非特异性切割的rna片段的读出,判断靶rna的存在,且具有较高的灵敏度。
目前,纳米孔技术可以检测的合适的核酸序列通常长度较短,因此从长rna序列上截取适合纳米孔分析的包含修饰位点的目标片段的样品制备方法是十分重要的。rnaseh酶是一种rna内切酶,其不能切割单链dna或单链rna,但可以切割dna与rna形成的双链。因此当已知靶rna的序列,拟利用纳米孔技术进行rna修饰检测时,可先设计dna序列,与包含修饰位点的目标rna片段的周围序列互补配对成双链,经rnaseh酶切割可得适合纳米孔检测长度的,包含修饰位点的目标rna片段。
rna的表观遗传修饰是rna调节基因表达的基础,rna经过化学修饰后会影响其活性、定位和稳定性,常见的rna表观遗传修饰包括m6a,m1a,m7g,poly(a)尾,u尾等。其中m6a是真核生物rna中最丰富的修饰类型,其可以调节rna的代谢,改变rna的折叠和结构,影响rna的成熟与降解,促进mrna的转录,影响细胞功能等等。m6a修饰后不影响其与t的配对,因此使用rt-pcr进行rna测序时会丢失修饰的信息。
技术实现要素:
本发明所要解决的技术问题是:提供突变aerolysin单体在检测rna碱基序列及rna修饰方面的应用,发现突变aerolysin单体制备的纳米孔可以检测mrna片段中的m6a等修饰,从混合物中将甲基化与未甲基化的rna链区分开,还可以用于检测靶rna的存在与否。
为了解决上述技术问题,本发明采用的技术方案如下:提供一种突变aerolysin单体在检测rna碱基序列及rna修饰方面的应用;所述突变aerolysin单体包含t232k/k238q两个氨基酸位点的突变。
优选的,所述突变aerolysin单体还包含n226q、g270k、s228k、t232k、k238q中至少一个的氨基酸位点的突变。
其中,所述aerolysin单体在检测rna碱基序列及其修饰方面的应用,其特征在于,所述rna修饰为rna上的碱基修饰,其包括但不限于m6a,m1a,m7g,poly(a)尾,u尾。
所述突变aerolysin单体在检测rna碱基序列的具体方法如下:(a)设计crrna序列,利用crispr-cas13a和crrna特异性识别包含碱基修饰的靶rna序列,并将长链rna序列切割成短的包含修饰碱基的短rna序列,(c)设计dna序列与靶rna片段的周围序列互补配对;(d)用核酸内切酶切割dna-rna双链,得到靶rna序列;(e)使用aerolysin单体制备的纳米孔检测靶rna片段的信号,从而确认靶rna的存在及其修饰。
其中,上述步骤(e)使用aerolysin单体制备的纳米孔检测靶rna片段的信号具体方法为:使所述靶rna片段相对于aerolysin单体制备的纳米孔中移动,测量所述短rna的穿孔电流信号,根据数学变换解析出该电流信号的瞬时频率从而表征短rna片段。
本发明的有益效果如下:
使用本发明所述突变aerolysin单体检测rna碱基序列及rna修饰,使用aerolysin单体制备的纳米孔检测靶rna片段的信号,其所述孔道已具有控制核酸移动速度的功能,无需现有纳米孔rna测序中所使用的聚合酶的参与,无需特殊化学修饰或荧光标记,通过纳米孔电信号即可在单分子水平上对样品中是否含有目的rna序列进行检测以及对修饰的rna链进行区分。
附图说明
图1为本发明的crispr-cas13a检测靶rna存在示意图。
图2为本发明的rnaseh切割dna-rna双链获取目标rna链示意图。
图3为本发明的aerolysin突变孔用于检测rna修饰示意图。包括t232kk238q,n226qk238q,n226qg270k,n226qs228kaerolysin突变孔。
图4a为本发明的实施例1所述rna样品相对于孔的移动的电流信号示意图,rna1序列:5’-cagacucuguagcg-3’和rna2序列:5’-cagm6acucuguagcg-3’。图4b为本发明的实施例2所述rna样品相对于孔的移动的电流信号示意图,rna3序列:5’-ggacu-3’)和rna4序列:5’-ggm6acu-3’。
具体实施方式
本文所提到的剪切酶均指crispr-cas13a,其用于纳米孔检测rna修饰的样品前处理,特异性识别靶rna序列并剪切,而非指纳米孔测序中使用的聚合酶,本发明中纳米孔检测方法采用无酶的方案,突变aerolysin纳米孔实现对rna的控速。下述是本发明技术方案中所用到的几个基本原理
一、基于crispr-cas13的纳米孔检测靶rna
本发明提供了用crispr-cas13a特异性识别样品中是否存在靶rna序列的纳米孔检测的方法。包含靶向靶rna的crrna的设计与合成,crispr-cas13a系统的特异性识别和剪切。
靶向含有碱基修饰位点的目标rna的crrna的设计原则如下:
(1)crrna包括间隔序列(spacer)和直接重复序列(directrepeat,dr),序列格式为:5’-与cas13a蛋白结合的直接重复序列-crrna间隔序列-3’,直接重复序列需要根据cas13a蛋白来确定,使其能够与选定的cas13a蛋白匹配并结合;
(2)crrna的间隔序列长度为22~28个碱基序列;
(3)crrna的直接重复序列大于24个碱基序列;
(4)crrna的直接重复序列应包含茎环(stemloop)结构;
(5)在crrna的间隔序列中间为seed区,与靶序列结合时不能发生错配。
crispr-cas13系统特异性识别靶rna。所述crispr-cas13a核酸酶结合crrna可以被目标核酸序列特异性激活,并剪切该序列。crispr-cas核酸酶还可以包括lbacas13,lbuc13a,lwacas13a,aspcas13a,bzocas13a,ccacas13a,aspcas13b,bzocas13b,ccacas13b,psmcas13b,pincas13b,pbucas13b,pgucas13b,pigcas13b,psacas13b,rancas13b,pspcas13b,escas13d,rspcas13d。该酶可以包括野生型、改造过的、密码子优化过的、进化过的、嗜热的、工程化的和/或一种以上cas蛋白的混合物。crispr-cas核酸酶优选地为lwacas13a。
二rnaseh酶剪切获得包含修饰位点的目标rna链
本发明提供了样品制备的方法,用于纳米孔检测rna的碱基修饰。该方法包含设计dna序列与目标rna片段的周围序列互补配对,rnaseh剪切dna-rna双链从而得到包含修饰位点的适合纳米孔检测的目标rna链。rnaseh起核酸内切酶的作用需要二价阳离子的参与,其包括mg2+,mn2+,切割产生5’端的磷酸基团和3’端的羟基。rnaseh剪切的dna-rna双链至少有6对碱基互补配对。所述的rnaseh酶可以包括1型和2型,1型包括细菌的rnaseh1和真核生物的rnaseh1,2型包括细菌的rnaseh2和rnaseh3,古细菌的rnaseh2及真核生物的rnaseh2。
三、表征靶rna
本发明提供了表征靶rna的方法。已知靶rna序列和碱基修饰发生的位置,截取碱基长度为14的rna链,使该碱基修饰位点位于从3’端开始数的第11个碱基。甲基化和未甲基化的rna截取方式相同。获得该混合物rna后使用aerolysin突变孔表征。
本发明样品制备非常简单,因为纳米孔能够检测单个rna分子,不需扩增靶rna或互补核酸。本发明通常不涉及聚合酶链式反应或逆转录。这大大减少了表征靶rna的工作流程。
四、膜
本发明的纳米孔需要插入膜中构成一个体系。膜优选地是两亲层。两亲层是由两亲性分子形成的两亲膜,所述两亲性分子如磷脂,具有亲水性和亲脂性两种特性。两亲分子可以是合成的或天然存在的。
下面结合说明书附图对本发明作进一步说明。
实施例1rna特征序列的检测
使用aerolysin单体制备的纳米孔对rna特征序列的表征:,待测rna序列为天然牛催乳素ncbinm_173953.2的3’utr序列的一段片段,其rna序列和crrna序列见表1。cas13a蛋白(2mg/ml,4μl)使用前用122.5μl储液重悬。将1μl待测rna样品,2μl酶缓冲溶液(400mmtrisph7.4),9.6μldepc处理水,2μlcas13a蛋白溶液,1μlcr-rna(10ng/μl),supernase核糖核酸抑制剂1μl,mgcl2(120mm)1μl混合反应30min。所有反应完成后,涡旋充分混合,在离心机中自旋,并在37°c的水浴中孵育30分钟。孵育后,将反应管放回冰上。最后,收集产物使用t232k/k238q突变纳米孔检测由cas13a酶特异性切割下来的片段,若检测到切割片段,则说明待测rna样品中含有靶rna。
表1待测靶rna,crrna和dna序列
实施例2rna修饰的检测
rnaseh剪切目标rna用于纳米孔rna碱基修饰检测:第一步,退火。将待测rna样品30μl(约100μm,含无碱基修饰和有碱基修饰序列)与30μl的dna1(100μm)和30μl的dna2(100μm)混合,95°c变性5min,后慢慢冷却到室温,形成dna-rna双链。第二步,将第一步得到的样品90μl(约30μm)与22.5μl的5×rnaseh酶缓冲溶液(500mmtris-hcl;750mmkcl;30mmmgcl2;100mmdtt)和1μl的rnaseh(5u/μl)混合,并在37°c的水浴中孵育20分钟,靶rna序列将被切割成rna1:5’-cagacucuguagcg-3’和rna2:5’-cagm6acucuguagcg-3’。反应完成后,在65°c的水浴中孵育20分钟,使rnaseh酶失活。第三步,使用hplc纯化剪切所得的包含碱基修饰位点的目标rna链。
使用t232k/k238qaerolysin突变单体制备的纳米孔检测上述制备获得的靶rna片段的碱基修饰:制备t232k/k238qaerolysin纳米孔,用于检测rna短片段的t232k/k238qaerolysin突变孔如图3所示,对aerolysin纳米孔β桶内232位点和238位点氨基酸进行突变,可以延长rna链在孔道内的停留时间,根据甲基化修饰和未甲基化修饰的rna链与纳米孔道内氨基酸残基的相互作用不同产生的纳米孔电信号不同来进行检测区分。具体原理如下:在室温下用胰蛋白酶酶切proaerolysin蛋白6小时,得到有活性的aerolysin蛋白。实验所用的缩醛树脂检测池由分别称为cis端和trans端的两个检测池组成,两个检测池中分别含有1ml的缓冲溶液(1.0m/3.0mkcl,10mmtris,ph8.0),该缓冲溶液由depc处理水配制以防止rna的降解,磷脂双分子层趴附在检测池中间50µm的小孔上,在cis端检测池中加入一定量的t232k/k238qaerolysin突变单体蛋白溶液,t232k/k238qaerolysin突变单体自组装成七聚体并插入到磷脂双分子层上形成纳米孔道,用于控制rna的移动速度,所有的纳米孔电化学分析实验温度均控制在20±2℃。将经rnaseh剪切得到的目标rna样品(体积为40μl,浓度为20μm)加入到cis端检测池中,检测不同电压下的电流信号。使用膜片钳放大器(axopatch200b)放大和采集aerolysin纳米孔道内的电流信号,并通过模数转换器(digidata1440a)转换成数字信号。采样率为100khz,滤波频率为5khz;通过clampex10.4软件(moleculardevices,forestcity,ca)获取电流信号并使用mosaic和origin软件对所得数据进行统计分析。仪器所采集到的每个数据点的电流值,包括的内容有:所处时间点下纳米孔道的电流基线、电流阻断以及电流噪音的综合数据。由于某些信号阻断时间较短,处理时被认为是噪声而丢失,可以通过计算阻断数据中的所有极小极大值点来进行信号的识别。
采用上述方法检测甲基化和未甲基化rna混合物的电流信号,其相对于孔的移动的电流轨迹如图4a所示,若样品中含有特异性的rna链,则可以用cas13a酶切割,产生纳米孔阻断电流信号,若样品中不含特异性的rna链,则不会被cas13a酶切割,不会产生纳米孔阻断电流信号。
实施例3本实例描述用于纳米孔rna特征序列表征和将其剪切成5个核苷酸长度(rna3:5’-ggacu-3’和rna4:5’-ggm6acu-3’)进行修饰检测的方法。
采用rna序列为m6a最常见的保守序列。基于crispr-cas13a检测靶rna是否存在:待测rna序列和crrna序列见表2。cas13a蛋白(2mg/ml,4μl)使用前用122.5μl储液重悬。将1μl待测rna样品,2μl酶缓冲溶液(400mmtrisph7.4),9.6μldepc处理水,2μlcas13a蛋白溶液,1μlcr-rna(10ng/μl),supernase核糖核酸抑制剂1μl,mgcl2(120mm)1μl混合反应30min。所有反应完成后,涡旋充分混合,在离心机中自旋,并在37°c的水浴中孵育30分钟。孵育后,将反应管放回冰上。最后,收集产物使用t232k/k238q突变纳米孔检测。由cas13a酶特异性切割下来的片段,若检测到切割片段,则说明待测rna样品中含有靶rna。.
表2待测靶rna,crrna和dna序列
rnaseh剪切目标rna用于纳米孔rna碱基修饰检测:第一步,退火。将待测rna样品20μl(约100μm,含无碱基修饰和有碱基修饰序列)与20μl的dna3(100μm)混合,95°c变性5min,后慢慢冷却到室温,形成dna-rna双链。第二步,将第一步得到的样品90μl(约30μm)与10μl的5×rnaseh酶缓冲溶液(500mmtris-hcl;750mmkcl;30mmmgcl2;100mmdtt)和0.5μl的rnaseh(5u/μl)混合,并在37°c的水浴中孵育20分钟,靶rna序列将被切割成rna3:5’-ggacu-3’和rna4:5’-ggm6acu-3’。反应完成后,在65°c的水浴中孵育20分钟,使rnaseh酶失活。第三步,使用hplc纯化剪切所得的包含碱基修饰位点的目标rna链。
采用t232k/k238qaerolysin突变纳米孔检测上述制备获得的靶rna片段的碱基修饰:制备t232k/k238qaerolysin纳米孔,包括将质粒proaerolysin进行t232k/k238q两个氨基酸位点的突变并将其表达和纯化,用于检测rna的t232k/k238qaerolysin突变孔如图3所示,用于检测rna的t232kk238qaerolysin突变孔如图3左上所示,第238位点带正电荷的赖氨酸突变成不带电荷的谷氨酰胺,延长核酸的过孔时间,第232位点的苏氨酸突变成带正电荷的赖氨酸,增加灵敏位点。其他突变孔如n226qk238q/同样起着延长时间,灵敏探测的作用。在室温下用胰蛋白酶酶切proaerolysin蛋白6小时,得到有活性的aerolysin蛋白。实验所用的缩醛树脂检测池由分别称为cis端和trans端的两个检测池组成,两个检测池中分别含有1ml的缓冲溶液(1.0m/3.0mkcl,10mmtris,ph8.0),该缓冲溶液由depc处理水配制以防止rna的降解,磷脂双分子层趴附在检测池中间50µm的小孔上,在cis端检测池中加入一定量的t232k/k238qaerolysin突变单体蛋白溶液,t232k/k238qaerolysin突变单体自组装成七聚体并插入到磷脂双分子层上形成纳米孔道,用于控制rna的移动速度,所有的纳米孔电化学分析实验温度均控制在20±2℃。将经rnaseh剪切得到的目标rna样品(体积为20μl,浓度为50μm)加入到cis端检测池中,检测不同电压下的电流信号。使用膜片钳放大器(axopatch200b)放大和采集aerolysin纳米孔道内的电流信号,并通过模数转换器(digidata1440a)转换成数字信号。采样率为100khz,滤波频率为5khz;通过clampex10.4软件(moleculardevices,forestcity,ca)获取电流信号并使用mosaic和origin软件对所得数据进行统计分析。仪器所采集到的每个数据点的电流值,包括的内容有:所处时间点下纳米孔道的电流基线、电流阻断以及电流噪音的综合数据。由于某些信号阻断时间较短,处理时被认为是噪声而丢失,可以通过计算阻断数据中的所有极小极大值点来进行信号的识别。
采用上述方法检测甲基化和未甲基化rna混合物的电流信号,其相对于孔的移动的电流轨迹如图4b所示,与未甲基化的rna相比,甲基化的rna阻断程度更大,阻断时间更长,可能是与孔道内r220区域的相互作用更强。
以上显示和描述了本发明的基本原理、主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的仅为本发明的优选例,并不用来限制本发明,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
- 还没有人留言评论。精彩留言会获得点赞!