一种3-烷硫基异噻唑衍生物及其合成方法
1.本发明涉及一种3-烷硫基异噻唑衍生物及其合成方法,属于化学有机合成技术领域。
背景技术:
2.3-烷硫基异噻唑衍生物是一类具有广泛生物活性的化合物,可用于中枢神经系统镇定剂、药物、除草剂、防蛀剂、防腐剂、杀菌剂等。因此,发展合成3-烷硫基异噻唑衍生物的新方法有着非常重要的意义。目前,有多种方法用于3-烷硫基异噻唑衍生物的制备,但是采用α-硫羰基-n,s-缩烯酮为原料合成3-烷硫基异噻唑衍生物的文献报道只有1种。相关制备方法为:α-硫羰基-n,s-缩烯酮先与碘单质反应,形成异噻唑五元环碘盐;再与碘化钾加热进行反应(j.org.chem.2002,67,5375)。然而,上述方法需要需要面临合成步骤复杂,条件较为苛刻,反应收率较低,还需要一定量的碘单质,使得其在3-烷硫基异噻唑衍生物合成中的应用受到了一定的限制。
3.鉴于采用α-硫羰基-n,s-缩烯酮为原料合成3-烷硫基异噻唑的合成方法步骤繁琐、条件苛刻且效率较低,因此,以易制备、具有结构多样性和多反应中心的α-硫羰基-n,s-缩烯酮为原料仅需一步氧化环化反应,通过调控α-硫羰基-n,s-缩烯酮中r1、r2、r2取代基,合成一系列不同结构的3-烷硫基异噻唑衍生物。
技术实现要素:
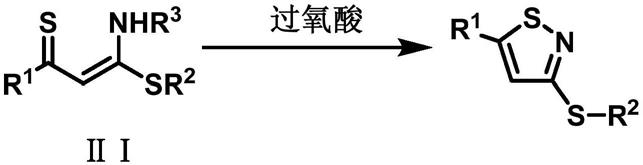
4.本发明的目的在于以易制备、具有结构多样性和多反应中心的α-硫羰基-n,s-缩烯酮ⅱ为原料一步实现了异噻唑环的构建,合成具有潜在药物活性的3-烷硫基异噻唑衍生物。
5.本发明提供一种3-烷硫基异噻唑衍生物,其分子结构式ⅰ如下:
[0006][0007]
r1选自甲基、芳基、萘环、呋喃环、噻吩环或环丙烷基;r2选自甲基、乙基、环丙烷基或芳基;其中芳基选自苯基、苯环上带有取代基的芳基,苯环上带有的取代基选自甲基、甲氧基、氟、氯、溴、碘、三氟甲基、硝基、氰基、羧基中的1-5种,取代基的个数为1-5个。
[0008]
本发明提供一种3-烷硫基异噻唑衍生物ⅰ的合成方法,以α-硫羰基-n,s-缩烯酮ⅱ为起始原料,过氧酸为促进剂,在加热或不加热条件下,在溶剂中自身发生氧化环化反应,一步生成3-烷硫基异噻唑衍生物ⅰ。
[0009]
α-硫羰基-n,s-缩烯酮ⅱ的分子结构式如下:
[0010][0011]
r1选自甲基、芳基、萘环、呋喃环、噻吩环或环丙烷基;r2选自甲基、乙基、环丙烷基或芳基;其中芳基选自苯基、苯环上带有取代基的芳基,苯环上带有的取代基选自甲基、甲氧基、氟、氯、溴、碘、三氟甲基、硝基、氰基、羧基中的1-5种,取代基的个数为1-5个;r3选自甲基、乙基、苄基、芳基、萘环、呋喃环、噻吩环、环丙烷基;
[0012]
合成路线如下述反应式所示:
[0013][0014]
其中:过氧酸促进剂选自过氧乙酸(ch3co2oh)、过氧三氟乙酸(cf3co2oh)、过氧化氢(h2o2)、过氧苯甲酸(phco2oh)、间氯过氧苯甲酸(m-clc6h4co2oh)、4-硝基过氧化苯甲酸(p-no2c6h4co2oh)、过氧苯乙酸(phch2co2oh)中的一种或二种以上,α-硫羰基-n,s-缩烯酮ⅱ与过氧酸的摩尔比为1:2.0-1:5.0;
[0015]
反应溶剂选自n,n-二甲基甲酰胺(dmf)、二甲基亚砜(dmso)、三氟甲苯、乙腈、甲苯(phme)、1,4-二氧六环、四氢呋喃(thf)中的一种或两种以上的混合物;α-硫羰基-n,s-缩烯酮ⅱ在反应溶剂中的摩尔浓度为0.05-1.0m;
[0016]
反应气氛为空气、氧气、氮气或氩气中的一种或两种以上;反应时间为0.1-48小时;反应温度为0-130℃。
[0017]
进一步地,在上述技术方案中,α-硫羰基-n,s-缩烯酮ⅱ生成ⅰ的反应中过氧酸最好是m-clc6h4co2oh。
[0018]
进一步地,在上述技术方案中,α-硫羰基-n,s-缩烯酮ⅱ生成ⅰ的反应最好在非质子非极性溶剂甲苯中进行。
[0019]
进一步地,在上述技术方案中,α-硫羰基-n,s-缩烯酮ⅱ生成ⅰ的反应最好在氮气气氛中进行。
[0020]
进一步地,在上述技术方案中,α-硫羰基-n,s-缩烯酮ⅱ生成ⅰ的反应最佳反应时间为1-3小时。
[0021]
进一步地,在上述技术方案中,α-硫羰基-n,s-缩烯酮ⅱ生成ⅰ的反应最佳反应温度是50-90℃。
[0022]
进一步地,在上述技术方案中,α-硫羰基-n,s-缩烯酮ⅱ生成ⅰ的反应中α-硫羰基-n,s-缩烯酮ⅱ与过氧酸的优选摩尔比为1:2.0。
[0023]
本发明以易制备、具有结构多样性和多反应中心的α-硫羰基-n,s-缩烯酮ⅱ为起始原料,过氧酸为促进剂,通过氧化环化反应,一步构建3-烷硫基异噻唑环,生成一系列3-烷硫基异噻唑衍生物,所得3-烷硫基异噻唑衍生物具有一定潜在药物活性。与已报道的以α-硫羰基-n,s-缩烯酮为原料合成异噻唑衍生物的方法相比较,本发明仅需一步反应、操作
简便、条件温和、合成反应效率高,收率在50%-88%,且产物具有很好的立体选择性及官能团多样性。本发明合成的3-烷硫基异噻唑骨架结构可以作为药物及多种化工用品结构的中间体。
[0024]
本发明具有以下优点:
[0025]
1)合成子α-硫羰基-n,s-缩烯酮ⅱ具有结构多样性,可以用来合成不同类型和结构的3-烷硫基异噻唑衍生物ⅰ。
[0026]
2)合成子ⅱ商业可得,成本低廉,易于工业化生产。
[0027]
3)3-烷硫基异噻唑衍生物ⅰ的合成反应使用价格较低相对无毒的rco2oh作为促进剂。
[0028]
4)3-烷硫基异噻唑衍生物ⅰ合成反应仅需一步构建异噻唑环,产物收率高,最高可达到88%。
[0029]
5)3-烷硫基异噻唑衍生物ⅰ合成反应条件较温和,温度范围为50-90℃。
[0030]
6)3-烷硫基异噻唑衍生物ⅰ产物有好的立体选择性,及官能团多样性,具有广泛的应用性。
[0031]
总之,本发明利用α-硫羰基-n,s-缩烯酮ⅱ的结构多样性与多反应中心来高效合成不同类型和结构的3-烷硫基异噻唑衍生物ⅰ,原料便宜易得,仅需一步反应,得到一系列3-烷硫基异噻唑衍生物结构,操作简便,条件温和,目标产物收率高。
具体实施方式
[0032]
在110℃下,在甲苯溶剂中,α-羰基-n,s-缩烯酮a与劳森试剂b反应生成α-硫羰基-n,s-缩烯酮ⅱ。式a中r1、r2、r3中定义同式ⅱ。
[0033][0034]
具体过程为:将α-羰基-n,s-缩烯酮a(2.0mmol)、劳森试剂b(1.0mmol)溶于3ml甲苯中,在110℃油浴中搅拌反应1min,tlc检测,原料α-羰基-n,s-缩烯酮a反应完全即停止反应。冷至室温后,减压下除去挥发组份,然后用硅胶柱层析分离(洗脱液为石油醚(60-90℃)/乙酸乙酯,v/v=50:1),得到目标产物ⅱ。目标产物通过核磁共振谱和高分辨质谱测定得到确认。
[0035]
下述实施例的原料2a、2b采用如下文献方法制备得到:
[0036]
z.q.liu,p.wu,y.he,t.yang,z.k.yu,adv.synth.catal.2018,360,4381-4392.
[0037]
通过下述实施例有助于进一步理解本发明,但本发明的内容并不仅限于此。
[0038]
实施例1
[0039]
[0040]
在手套箱中,依次称取1-甲硫基-1-苄胺-1-丁烯-3-苯基-3-硫酮2a(0.3mmol)、间氯过氧苯甲酸(0.60mmol)于25ml schlenk反应瓶中,在氮气下,加入甲苯2ml,放入60℃的油浴中反应2小时。反应结束后,将混合物冷却至室温,减压下除去挥发组份,然后用硅胶柱层析分离(洗脱液为石油醚(60-90℃)/乙酸乙酯,v/v=20:1),得到黄色液体目标产物1a(50mg,收率80%)。目标产物通过核磁共振谱测定得到确认。
[0041]
化合物表征数据
[0042]
3-甲硫基-5-苯基异噻唑衍生物(1a),黄色液体.1h nmr(400mhz,cdcl3)δ7.55(dd,j=7.7,2h,1.8hz),7.42(m,3h),7.15(s,1h),2.69(s,3h).
13
c{1h}nmr(100mhz,cdcl3)δ167.57,163.97,130.66,129.80,129.28,126.63,118.17,14.17.
[0043]
实施例2
[0044][0045]
在手套箱中,依次称取1-甲硫基-1-苄胺-1-丁烯-3-间溴苯基-3-硫酮2b(0.3mmol)、间氯过氧苯甲酸(0.60mmol)于25ml schlenk反应瓶中,在氮气下,加入甲苯2ml,放入60℃的油浴中反应2小时。反应结束后,将混合物冷却至室温,减压下除去挥发组份,然后用硅胶柱层析分离(洗脱液为石油醚(60-90℃)/乙酸乙酯,v/v=20:1),得到黄色液体目标产物1b(65mg,收率75%)。目标产物通过核磁共振谱测定得到确认。
[0046]
化合物表征数据
[0047]
3-甲硫基-5-间溴苯基异噻唑衍生物(1b),黄色液体.1h nmr(400mhz,cdcl3)δ7.69(t,j=1.8hz,1h),7.52(m,1h),7.46(m,1h),7.29(m,1h),7.13(s,1h),2.68(s,3h).
13
c{1h}nmr(100mhz,cdcl3)165.76,164.31,132.69,132.62,130.82,129.54,125.33,123.36,118.85,14.22.
[0048]
实施例3
[0049]
反应步骤与操作同实施例1,与实施例1不同之处在于,2a与的m-clc6h4co2oh摩尔比为1:1.5。停止反应,经后处理得到目标产物1a(43mg,收率70%)。
[0050]
实施例4
[0051]
反应步骤与操作同实施例1,与实施例1不同之处在于,phme改为dmf。停止反应,经后处理得到目标产物1a(38mg,收率62%)。
[0052]
实施例5
[0053]
反应步骤与操作同实施例1,与实施例1不同之处在于,dmf改为dmso。停止反应,经后处理得到目标产物1a(40mg,收率65%)。
[0054]
实施例6
[0055]
反应步骤与操作同实施例1,与实施例1不同之处在于,m-clc6h4co2oh改为ch3co2oh。停止反应,经后处理得到目标产物1a(33mg,收率53%)。
[0056]
实施例7
[0057]
反应步骤与操作同实施例1,与实施例1不同之处在于,m-clc6h4co2oh改为phco2oh。停止反应,经后处理得到目标产物1a(38mg,收率61%)。
[0058]
实施例8
[0059]
反应步骤与操作同实施例1,与实施例1不同之处在于,n2改为o2。停止反应,经后处理得到目标产物1a(34mg,收率55%)。
[0060]
实施例9
[0061]
反应步骤与操作同实施例1,与实施例1不同之处在于,n2改为空气。停止反应,经后处理得到目标产物1a(37mg,收率60%)。
[0062]
实施例10
[0063]
反应步骤与操作同实施例1,与实施例1不同之处在于,60℃改为40℃。停止反应,经后处理得到目标产物1a(31mg,收率51%)。
[0064]
本发明方法原料易得,仅需一步反应,操作简便,合成反应条件温和、反应效率高,其官能团具有多样性。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1