一类含紫精单元的离子型铱(III)配合物及其制备方法和应用与流程
本发明属于有机光电功能材料领域,更具体地说,涉及一类含紫精单元的离子型铱(iii)配合物及其制备方法和应用。
背景技术:
忆阻器,一种非易失性存储器,它是具有记忆功能的非线性电阻元件,是继电阻、电感、电容之后的第四种无源电路元件,主要是利用某些薄膜材料在电激励的作用下会出现不同电阻状态(高、低阻态)的转变现象来进行数据的存储。由于具有超小的尺寸,极快的擦除/写入速度,超高的擦除/写入寿命,多电阻开关特性以及良好的互补金属氧化物半导体(cmos)兼容性,因此被认为是存储器和神经突触电子仿生器件的重要候选者。
作为常规无机半导体活性材料的替代品,由于有机和含金属的材料易于结构修饰,良好的可扩展性,高柔性和良好的可加工性,非常适用于构建忆阻器。其中,过渡金属配合物由于其独特的电子性能,如丰富的激发态性能,优异的电亲和性,良好的氧化还原可逆性和对电刺激的高敏感性而受到了广泛的关注,这有助于提高电子存储器件的稳定性并实现多状态存储。因此,设计、合成这类具有电阻特性的金属配合物具有很重要的价值和意义。
技术实现要素:
针对上述存在的问题,本发明旨在提供一类含紫精单元的的离子型铱(iii)配合物及其制备方法,利用紫精丰富的氧化还原态、良好的氧化还原可逆性和优异的接受电子能力及离子型铱(iii)配合物的温和的合成条件、丰富的电荷转移激发态,良好的电亲和性,优异的氧化还原可逆性和对电刺激的敏感性,通过将紫精单元引入n^n配体后与不同的c^n配体结合制成了一类含紫精单元的离子型铱(iii)配合物。此类离子型铱(iii)配合物具有比单个紫精单元和不含紫精单元的或其它含紫精单元的离子型铱(iii)配合物更优异的性质,如更多的氧化还原态、更丰富的电荷转移激发态和对电刺激更高的敏感性,非常有利于忆阻行为的实现。通过器件结构优化,制备出性能优异的忆阻器,并且通过电压调控,实现了多态存储,这大大提高了信息存储的能力。而且,这种优异的忆阻特性在神经突触仿生方面表现出巨大的应用潜力。
为了实现上述目的,本发明采用以下技术方案:
一类含紫精单元的离子型铱(iii)配合物,该类铱(iii)配合物由金属中心铱原子、不同的抗衡阴离子、不同的c^n配体及n^n配体组成。
一类含紫精单元的的离子型铱(iii)配合物包括ir-phen-x-,ir-bpy-x-两种,结构式分别为:
其中,x-为下列中的任意一个:x-=pf6-,bf4-,i-,br-,cl-,tfsi-;
其中,c^n配体的结构选自下列结构中的一个:
一类含紫精单元的离子型铱(iii)配合物合成路线如下:
一类含紫精单元的的离子型铱(iii)配合物的具体合成步骤为:
(1)化合物3的制备:将4,4'-联吡啶和1-氯-2,4-二硝基苯在乙腈、乙醇或丙酮溶剂中回流反应72h以内,反应结束后抽滤,将滤液旋干并用乙醚洗涤三次,真空干燥得化合物3;
(2)化合物1和7的制备:将化合物1,10-菲罗啉和2,2-联吡啶分别与浓硫酸和浓硝酸混合溶液在150~200℃下反应6h以内,反应结束后冷却至室温,将反应液倒入冰水中并调其ph为酸性至沉淀析出。过滤,用去离子水洗涤沉淀三次,真空干燥得化合物1和7;
(3)化合物2和8的制备:化合物1和7分别与催化剂钯碳,水合肼在乙醇中回流搅拌4~12h;反应结束后趁热过滤除去钯碳,旋蒸除去乙醇和过量的水合肼得化合物2和8;
(4)化合物4和9的制备:化合物2和8分别与化合物3在乙醇和去离子水的混合溶剂中回流反应24~84h;冷却至室温,旋蒸除去溶剂,加入少量的甲醇将固体完全溶解,再加入大量的乙酸乙酯和丙酮沉降,过滤,用乙酸乙酯和丙酮的混合溶剂洗涤三次得化合物4和9;
(5)化合物5和10的制备:化合物4和9分别和六氟磷酸盐在醇溶剂或去离子水中室温搅拌4~12h;反应结束后,过滤,用去离子水洗涤三次得化合物5和10;
(6)化合物6和11的制备:化合物5和10分别与不同金属二氯桥在甲醇和二氯甲烷的混合溶剂中在氮气氛围下回流反应12~36h;反应结束后,加入六氟磷酸钾室温搅拌4~12h;过滤除去未反应的六氟磷酸盐,并用甲醇和二氯甲烷的混合溶剂洗涤三次;旋干滤液,加入少量甲醇将固体完全溶解并加入大量乙醚重结晶得化合物6和11。
(7)化合物ir-phen-pf6-和ir-bpy-pf6-的制备:化合物6和11分别与碘甲烷在乙腈溶剂中和氮气氛围下于40~50℃反应12~36h;反应结束后,旋干除去溶剂;将固体溶解于甲醇和去离子水的混合溶剂中,加入六氟磷酸盐室温搅拌4~12h;反应结束后,过滤,除去未反应的六氟磷酸盐;将固体用少量二氯甲烷溶解并加入大量正己烷重结晶得化合物ir-phen-pf6-和ir-bpy-pf6-。
(7)化合物ir-phen-x-和ir-bpy-x-的制备:化合物ir-phen-pf6-和ir-bpy-pf6-分别与nabf4、ki、nabr、nacl或litfsi在乙腈和去离子水的混合溶剂中室温搅拌4~12h,旋干重结晶得化合物ir-phen-x-和ir-bpy-x-。
本发明提供的一类含紫精单元的离子型铱(iii)配合物,具有多个氧化还原态,丰富的电荷转移激发态和极好的电子接受能力等优点,具有优异的忆阻行为,可以通过器件结构优化制成忆阻器和备神经突触电子仿生器件。。
本发明提供的一类含紫精单元的离子型铱(iii)配合物,可用作电致变色和电致变色发光双功能材料,通过掺杂电解质和器件结构优化制成电致变色和电致变色发光双功能器件。
本发明所述的一类含紫精单元的离子型铱(iii)配合物ir-phen-x-和ir-bpy-x-混合,或两种中任何一种与对不同电压响应的紫精材料相混合,在没有加压的情况下对数据进行保护,施加不同的电压可以读取不同的信息,从而应用于信息的记录-擦除,加密-解密与防伪等。
本发明提供的一类含紫精单元的离子型铱(iii)配合物,可作为颜色指示剂,与超级电容器、电池等储能器件相连用来肉眼观察它们的充放电状态形成电池、电存储器或超级电容器。
本发明所述的一类含紫精单元的离子型铱(iii)配合物,具有良好的电子受体,可以涂于电极表面,应用于电极修饰。
本发明的有益效果是:具有丰富的氧化还原态、良好的电子亲和性、优异的氧化还原可逆性和对电刺激的高敏感性的金属有机材料非常有利于忆阻特性的实现,可以制备成忆阻器。本发明将紫精单元引入n^n配体中,制成了一类含紫精单元的离子型铱(iii)配合物。此类离子型铱(iii)配合物具有比紫精单元、不含紫精单元或其他含紫精单元的离子型铱(iii)配合物更优异的性质,即更多的氧化还原态、更丰富的电荷转移激发态、更优异的氧化还原可逆性和对电刺激更高的敏感性,实现了电阻特性的转换;通过器件结构优化,制备出了具有多阻态的忆阻器,即低开关阈值电压,高on/off电流比和在低读取电压下具有较长的保持时间。以化合物ir-phen-pf6-为例,它具有金属到配体的电荷转移(1mlct和3mlct,见图1)、配体到配体的电荷转移(llct,见图1)、六个氧化还原态和优异的氧化还原可逆性(见图2);以化合物ir-phen-pf6-为活性层制成的忆阻器具有0.5v的低开关阈值电压(见图3),约105/103/102/1的高on1/on2/on3/off电流比(见图4)和在0.05v低读取电压下具有2000s的较长的保持时间(见图5)。这种优异的忆阻特性在神经突触电子仿生器件方面表现出巨大的应用潜力。而且,该类离子型铱(iii)配合物还可以应用于光学显示和能源存储等方面。除此之外,紫精类衍生物原料易获得、合成简单及制作成本低且该类离子型铱(iii)配合物合成条件也比较温和,易于实现忆阻器的商业生产化。
附图说明
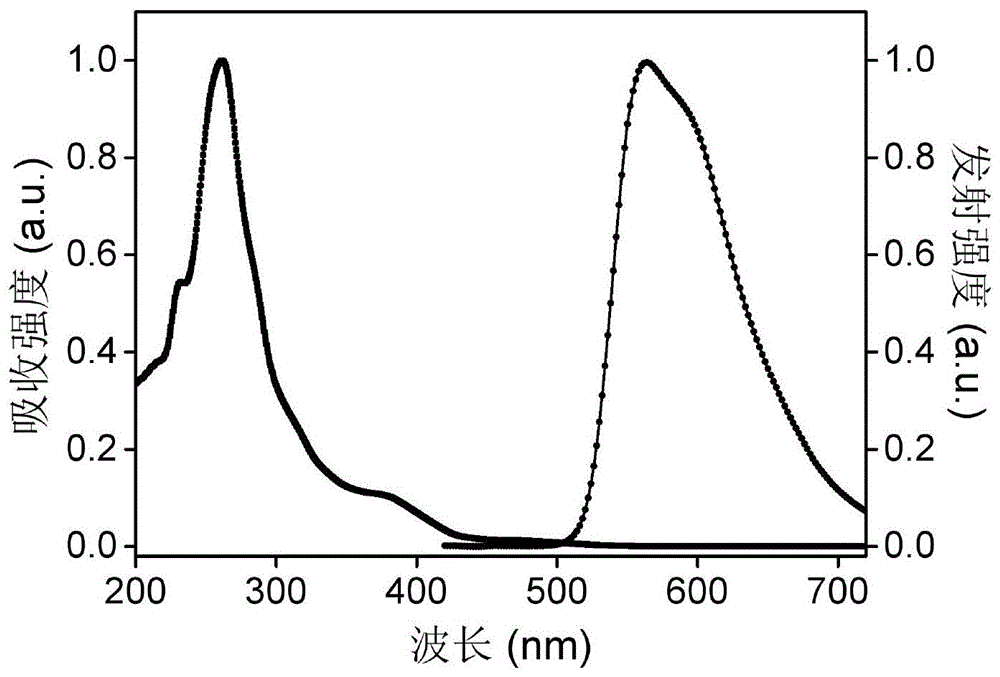
图1.实施例3中ir-phen-pf6-的紫外-可见吸收和发射光谱;
图2.实施例4中ir-phen-pf6-的循环伏安图;
图3.实施例5中ir-phen-pf6-的电流-电压(i-v)特性变化图;
图4.实施例6中基于ir-phen-pf6-的忆阻器的多态存储图;
图5.实施例7中基于ir-phen-pf6-的忆阻器的滞留时间曲线图。
具体实施方式
以下实施例进一步说明本发明的内容,但不应理解为对本发明的限制。在不背离本发明实质的情况下,对本发明方法、步骤或条件所作的修改和替换,均属于本发明的范围。若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段。
化合物ir-phen-x-和ir-bpy-x-具有相似的优异性质,即具有多个氧化还原态、丰富的电荷转移激发态、优异的氧化还原可逆性和对电刺激更高的敏感性。其中,ir-phen-pf6-和ir-bpy-pf6-具有相似的合成步骤、相似的光物理特性和忆阻特性,下面以ir-phen-pf6-为例进行详细说明。
实施例1:ir-phen-pf6-的制备
(1)化合物3的制备:4,4'-联吡啶(154mg,1mmol)和1-氯-2,4-二硝基苯(242mg,1.2mmol)在无水乙腈中回流反应72h,反应结束后,冷却至室温,抽滤,用乙腈洗涤三次,将滤液旋干并真空干燥得化合物3。产率:71%。1hnmr(400mhz,d2o)δ9.30(d,j=2.5hz,1h),9.16(d,j=7.1hz,2h),8.84(dd,j=8.7,2.5hz,1h),8.76-8.73(m,2h),8.59(d,j=7.1hz,2h),8.18(d,j=8.7hz,1h),7.95-7.91(m,2h).13cnmr(100mhz,d2o)δ150.7,148.8,143.9,139.9,134.4,133.2,132.6,123.7,121.0。
(2)[ir(ppy)2-μ-cl]2的制备:苯基吡啶(2.2mmol,341mg)和水合三氯化铱(1.0mmol,317mg)在乙二醇乙醚和去离子水的混合溶剂(体积比:3:1)中和氮气氛围下回流反应24h;反应结束后,冷却至室温,加去离子水,过滤,用去离子水洗涤三次并真空干燥得[ir(ppy)2-μ-cl]2。
(2)化合物1的制备:将1,10-菲罗啉(1g,5.56mmol)溶于6ml的浓硫酸中并加热至160℃,随后逐滴加入6ml的发烟硝酸反应3h;反应结束后,将混合液倒入冰水中,加入饱和的氢氧化钠溶液至沉淀析出;过滤并用去离子水洗涤三次,真空干燥得化合物1。1hnmr(400mhz,dmso-d6)δ9.41(dd,j=4.8,1.6hz,1h),9.36(dd,j=4.6,1.5hz,1h),9.26(s,1h),9.20-9.15(m,2h),8.23(ddd,j=11.6,8.4,4.7hz,2h).13cnmr(100mhz,dmso-d6)δ158.1,153.9,150.5,149.9,138.5,137.6,136.9,127.5,124.8,124.2,123.7,121.5。
(3)化合物2的制备:化合物1(225mg,1mmol)、质量分数为10%的钯碳(10mg)和水合肼(1ml)在乙醇中回流反应5h;反应结束后,过滤除去钯碳,旋蒸除去溶剂得化合物2。产率:98%。1hnmr(400mhz,dmso-d6)δ9.05(dd,j=4.3,1.6hz,4h),8.72-8.66(m,8h),8.06(dd,j=8.1,1.7hz,4h),7.75(dd,j=8.3,4.3hz,4h),7.52(dd,j=8.1,4.2hz,4h),6.87(s,4h),6.17(s,8h),3.44(s,15h),1.22(s,1h).13cnmr(100mhz,dmso-d6)δ149.9,148.7,146.6,145.1,138.4,135.0,132.9,128.5,124.1,121.5,120.6,121.0。
(4)化合物4的制备:化合物2(2.2mmol,429mg)和化合物3(2.0mmol,786mg)在80%的乙醇溶剂中回流反应24h;冷却至室温,旋蒸除去溶剂,加入少量甲醇将固体完全溶解,再加入大量的乙酸乙酯和丙酮的混合溶剂沉降得化合物4。产率:83.0%.1hnmr(400mhz,dmso-d6)δ9.65(d,j=6.8hz,2h),9.31(ddd,j=12.2,4.2,1.4hz,2h),9.02-8.96(m,4h),8.70(dd,j=8.0,1.6hz,1h),8.66(s,1h),8.23(dd,j=4.6,1.4hz,2h),8.11(dd,j=8.2,1.4hz,1h),7.99(dd,j=8.0,4.4hz,1h),7.89(dd,j=8.4,4.2hz,1h).13cnmr(100mhz,dmso-d6)δ150.0,152.8,151.9,151.6,147.5.145.9,145.5,141.4,138.1,136.8,131.1,126.9,126.4,126.3,125.1,124.9,123.9,122.7。
(5)化合物5的制备:将化合物4(1.0mmol,640mg)溶解在甲醇中并搅拌,逐滴加入饱和的六氟磷酸钾(5.0mmol,920mg)溶液至不再产生沉淀;将混合物在室温下搅拌12h,过滤,用去离子水洗涤三次,真空干燥得化合物5。产率:90.0%.1hnmr(400mhz,dmso-d6)δ9.70-9.65(m,2h),9.36-9.23(m,2h),8.99(t,j=6.4hz,4h),8.66(d,j=8.0hz,1h),8.62(s,1h),8.23(d,j=4.8hz,2h),8.09-8.03(m,1h),7.97(dd,j=8.0,4.4hz,1h),7.88-7.81(m,1h).13cnmr(100mhz,dmso-d6)δ154.9,152.8,151.9,151.7,147.6,146.2,145.7,141.2,137.9,136.9,131.2,126.9,126.4,126.3,125.1,124.8,124.1,122.6.19fnmr(376.5mhz,dmso-d6)δ-70.13(d,jf-p=711.59hz).
(6)化合物12的制备:化合物5(1.1mmol,480mg)和[ir(ppy)2-μ-cl]2(1.0mmol,1072mg)在二氯甲烷和甲醇的混合溶剂(体积比:3:1)中和氮气氛围下回流反应24h;反应结束后,冷却至室温,加入六氟磷酸钾(5.0mmol,920mg)搅拌4h;过滤除去未反应的六氟磷酸钾;旋干滤液,加入少量的二氯甲烷将其完全溶解再加入大量的乙醚沉降得化合物12。产率:51.0%.1hnmr(400mhz,dmso-d6)δ9.63(d,j=6.0hz,1h),9.56(d,j=6.6hz,1h),9.09-8.96(m,6h),8.58-8.53(m,1h),8.43-8.40(m,1h),8.39-8.35(m,1h),8.34-8.28(m,2h),8.25-8.20(m,3h),8.14(dd,j=8.8,5.2hz,1h),7.99(dd,j=7.2,2.8hz,2h),7.93(t,j=8.0hz,2h),7.60(d,j=5.6hz,1h),7.48(d,j=6.0hz,1h),7.13-7.04(m,4h),7.05-6.95(m,2h),6.32(t,j=6.8hz,2h).13cnmr(100mhz,dmso-d6)δ167.3,155.4,153.3,152.4,151.7,150.0,149.6,149.3,147.5,147.4,147.1,146.8,144.4,141.1,140.3,139.4,138.2,134.4,131.7,130.9,129.5,129.0,128.6,128.1,127.4,126.4,125.7,124.5,124.3,123.1,122.6,120.6.19fnmr(376.5mhz,dmso-d6)δ-70.09(d,jf-p=711.21hz).maldi-tof-ms(m/z):calcd.forc44h31irn6p2f12,1126.15;found,681.21.
(7)配合物ir-phen-pf6-的制备:化合物12(0.5mmol,420mg)和碘甲烷(8.0mmol,0.5ml)在乙腈溶剂和氮气氛围下于43℃反应24h;反应结束后,将反应液旋干除去未反应的碘甲烷和乙腈溶剂;随后,将固体溶解在甲醇中并搅拌,逐滴加入饱和的六氟磷酸钾(2.5mmol,460mg)溶液至不再产生沉淀;将混合物在室温下搅拌12h,过滤,用去离子水洗涤三次,真空干燥后用少量二氯甲烷将其完全溶解,再加入大量正己烷沉降得配合物ir-phen-pf6-。产率:90.0%.1hnmr(400mhz,dmso-d6)δ9.76(dd,j=30.4,6.4hz,1h),9.46-8.83(m,8h),8.57-8.51(m,1h),8.43(dd,j=5.2,1.2hz,1h),8.41-8.36(m,1h),8.32(t,j=8.0hz,2h),8.25(dd,j=8.0,5.2hz,1h),8.16(dd,j=8.8,5.2hz,1h),8.05-7.89(m,4h),7.61(d,j=5.6hz,1h),7.49(d,j=6.0hz,1h),7.19-6.94(m,7h),6.40-6.29(m,2h),4.50(s,3h).13cnmr(100mhz,dmso-d6)δ167.4,153.5,152.5,151.7,149.8,149.7,149.5,149.3,148.3,148.2,148.0,147.4,147.1,146.8,144.5,144.4,139.4,138.2,134.3,131.8,130.9,129.5,129.0,128.6,128.3,127.6,127.2,126.8,125.7,124.5,124.3,123.1,120.6,48.7.19fnmr(376.5mhz,dmso-d6)δ-70.09(d,jf-p=711.59hz).maldi-tof-ms(m/z):calcd.forc45h34irn6p3f18,1186.14;found,851.27.
实施例2:用ir-phen-pf6-制成的忆阻器的具体操作步骤如下:
(1)通过磁控溅射在350μm厚的sio2/si衬底上沉积带状钨(w,厚80nm)底部电极;
(2)将配合物ir-phen-pf6-的乙腈溶液(10mgml-1)以1000rpm的速度旋涂30s到带有sio2/si衬底的w底部电极上;
(3)通过磁控溅射将ag(厚度为100nm)沉积在ir-phen-pf6-活性层的表面上作为顶电极。
实施例3:ir-phen-pf6-的吸收和发射光谱测试
本发明采用的光谱测试浓度为10μm,测试溶剂为乙腈。测发射光谱时,ir-phen-pf6-的激发波长为405nm.
ir-phen-pf6-的吸收和发射光谱如图1所示。ir-phen-pf6-有三个吸收峰,分别在261,380and475nm左右。261nm左右的强吸收带归属于c^n和n^n配体的π→π*电子跃迁。380and475nm左右的弱吸收带分别归属于配体到配体的电荷转移(llct)和金属到配体的电荷转移(mlct)。ir-phen-pf6-的发射峰在572nm左右,发橙光,这可能是由于金属到配体的电荷转移(3mlct)激发态引起的。
实施例4:ir-phen-pf6-的循环伏安测试
ir-phen-pf6-的循环伏安测试采用三电极体系,钯碳电极为工作电极、ag/agno3为参比电极、铂丝电极为对电极。电解质是四丁基六氟磷酸铵的乙腈溶液(0.1m)。扫描速度是100mv·s-1。
ir-phen-pf6-的循环伏安图如图2所示。由图可知,此化合物共有两对可逆的氧化还原峰、四个非可逆的氧化还原峰。其中,处于负电位的两对可逆的氧化还原峰对应于紫精基团上两个氮原子分别得一个电子和两个电子的还原电位,其电位分别为ered1=-0.53v,ered2=-0.90v;处于负电位的两个非可逆的氧化还原峰分别对应于1,10-菲罗啉上的两个氮原子的两次失电子还原过程,其电位分别为ered3=-1.60v,ered4=-2.29v;处于正电位的两个非可逆的氧化还原峰对应于金属中心ir(iv/iii)的氧化,其电位分别为eox1=1.12,eox2=1.76。
实施例5:基于ir-phen-pf6-的忆阻器的电阻特性的测试
基于ir-phen-pf6-的忆阻器的电阻特性曲线通过吉时利4200a-scs半导体分析仪测试获得。基于ir-phen-pf6-的忆阻器的电流-电压(i-v)特性变化图如图3所示。在逆时针电压扫描0→0.8→0→-0.8→0v下,获得了i-v特性的收缩磁滞回线,表明配合物ir-phen-pf6-具有良好的电阻行为。
实施例6:基于ir-phen-pf6-的忆阻器的多态存储测试
基于ir-phen-pf6-的忆阻器的多态存储变化图如图4所示。在0.8v的电压下,将忆阻器从hrs(“off”状态)调节至为lrs(“on1”状态)。通过调控负压范围(-0.5~-1.4v),许多不同的hrs通过“on1”状态切换。结果表明,通过控制reset电压的幅度可以很好地调制忆阻器的电阻状态。
实施例7:基于ir-phen-pf6-的忆阻器的滞留时间测试
基于ir-phen-pf6-的忆阻器的滞留时间变化图如图5所示。在读出电压为0.05v的“off”,“on1”,“on2”和“on3”状态下测试了忆阻器的数据保持能力。在读出测试期间,在四种状态下均未观察到电流的明显下降,实现了高的off/on1/on2/on3电流比(1/105/103/102),这表明忆阻器具有出色的保持力。
- 还没有人留言评论。精彩留言会获得点赞!