一种肟菌酯的制备方法与流程
1.本发明属于有机化合物制备技术领域,具体涉及一种肟菌酯的制备方法。
背景技术:
2.肟菌酯属于甲氧基丙烯酸类杀菌剂,具有高效、广谱、保护和治疗活性好、内吸活性高、耐雨水冲刷性好、持效期长等特性,对几乎所有的真菌纲病害如白粉病、锈病、颖枯病、网斑病、霜霉病、稻瘟病等均有良好的活性,其在土壤、水中可快速降解,对作物十分安全。肟菌酯适用于水稻、玉米、大豆等大宗作物和果蔬等经济作物,应用十分广泛。
3.肟菌酯(trifloxystrobin,简称tfs)的化学名称为(2z)-2-甲氧基亚氨基-2-(2-((1-(3-(三氟甲基)苯基)亚乙基氨基)氧甲基)苯基)乙酸甲酯,结构式如下:由于肟菌酯的应用范围广,需求量很大,因此,其制备工艺的研发也成为农药化学领域的研究热点。
4.us5221762公开了以邻甲基苯甲酸为原料,与亚硫酰氯回流得邻甲基苯甲酰氯,与氰化钾反应得邻甲基苯甲酰氰,然后水解、酯化、肟化、溴化得2-溴甲基-α-甲氧亚胺基苯乙酸甲酯,合成路线如下:
[0005][0006]
该方法以剧毒的氰化物作为原料,溴化反应收率低、选择性较差,而且产物不易提纯,从而影响最后一步缩合反应得到的粗品含量,导致目标产物的纯化困难,不利于规模化生产。
[0007]
cn103787916a公开了一种肟菌酯的制备方法,该方法以间三氟甲基苯乙酮肟与(e)-2-(2'-溴甲基苯基)-2-羰基乙酸甲酯-o-甲基酮肟为原料,在相转移催化剂的作用下进行反应;该方法与传统工艺相比,反应速度有所提升,但肟菌酯的收率为80%左右,产物收率不太理想。
[0008]
cn105294490a公开的肟菌酯合成方法中,首先以3-异色满酮和亚硝基叔丁酯在甲醇钠的催化下反应生成(e)-3-酮-4-(亚氨基)异色满,经柱色谱分离后,在碱性条件下和硫
酸二甲酯进行甲基化反应,得到化合物(e)-3-酮-4-(甲氧亚氨基)异色满;然后在甲醇的存在下与氯化亚砜反应,生成(e)-2-氯代甲基-α-甲氧亚胺基苯乙酸甲酯,最后与(e)-间三氟甲基苯乙酮肟在碱性条件下反应,得到肟菌酯。该方法的肟化反应和酯化反应中使用亚硝酸叔丁酯和硫酸二甲酯,原子经济性差,导致含叔丁醇、硫酸和甲醇的废水多,难以分离处理;而且氯化亚砜的使用导致反应生成的二氧化硫和氯化氢尾气多,不易吸收分离;同时,该方法中需要多次柱层析进行产物的分离,不适合工业化生产,最后一步肟菌酯的合成收率也不高。
[0009]
因此,开发一种原料毒性低、无需溴化反应、原子经济性高的工艺路线,以实现肟菌酯的高收率和高纯度制备,是本领域的研究重点。
技术实现要素:
[0010]
针对现有技术的不足,本发明的目的在于提供一种肟菌酯的制备方法,所述制备方法包括肟化反应、醚化反应、氯化反应、酯化反应和缩合反应五个步骤,反应条件温和,原子经济性高,无需进行复杂的中间体分离,三废产生量少,能够获得高纯度、高收率的肟菌酯,适合大规模的工业化生产。
[0011]
为达此目的,本发明采用以下技术方案:
[0012]
本发明提供一种肟菌酯的制备方法,所述制备方法包括如下步骤:
[0013]
(1)3-异色满酮、有机金属醇盐与亚硝酸甲酯进行肟化反应,得到3-异色满酮-4-亚硝酸金属盐,反应式如下:
[0014][0015]
其中,r1选自c1~c5直链或支链烷基,包括c1、c2、c3、c4或c5的直链或支链烷基,示例性地包括但不限于:甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、异戊基或新戊基等。
[0016]
m选自钾或钠。
[0017]
(2)将步骤(1)得到的3-异色满酮-4-亚硝酸金属盐与一氯甲烷进行醚化反应,得到3-酮-4-(甲氧亚氨基)异色满,反应式如下:
[0018][0019]
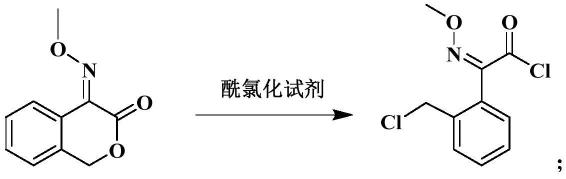
(3)将步骤(2)得到的3-酮-4-(甲氧亚氨基)异色满与酰氯化试剂进行氯化反应,得到2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酰氯;所述酰氯化试剂选自光气、双光气或固体光气中的任意一种或至少两种的组合;反应式如下:
[0020][0021]
(4)步骤(3)得到的2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酰氯与甲醇发生酯化反应,得到2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯,反应式如下:
[0022][0023]
(5)将步骤(4)得到的2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯与间三氟甲基苯乙酮肟进行缩合反应,得到所述肟菌酯,反应式如下:
[0024][0025]
本发明提供的制备方法包括肟化反应、醚化反应、氯化反应、酯化反应和缩合反应五个工艺步骤,通过亚硝酸甲酯和一氯甲烷引入甲氧胺基,亚硝酸甲酯的活性高,使用有机金属醇盐规避了水分引入,反应生成肟酸金属盐;步骤(1)生成的产物无需分离可以直接进入步骤(2)与一氯甲烷反应,原子经济性好,实现两步反应耦合,便于实现工业化。步骤(3)中以光气、双光气或固体光气作为酰氯试剂进行氯化反应,避免使用氯化亚砜产生的尾气导致氯化氢和二氧化硫难以分离、碱吸收导致混盐产生、固废量大等弊端。
[0026]
所述制备方法以3-异色满酮为起始原料,经过肟化、醚化、氯化和酯化四步得到2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯,原子经济性好,收率高,总收率达到85%,纯度≥97%,而且整个过程的中间体无需分离,三废少,适合工业化生产。酯化反应得到的产物与间三氟甲基苯乙酮肟进行缩合,即可得到肟菌酯,由于2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯的纯度高,因此缩合生成肟菌酯的收率达到96%以上,且含量高,便于提纯。本发明所述制备方法中,整个工艺路线的反应条件温和,不涉及剧毒性原料,无需复杂的中间产物处理步骤,三废产生量少,能够获得高纯度、高收率的肟菌酯。
[0027]
优选地,步骤(1)所述有机金属醇盐选自甲醇钠、甲醇钾、乙醇钠、乙醇钾、叔丁醇钠或叔丁醇钾中的任意一种或至少两种的组合,进一步优选为甲醇钠。
[0028]
优选地,步骤(1)所述3-异色满酮与有机金属醇盐的摩尔比为1:(1.2~1.5),例如可以为1:1.21、1:1.23、1:1.25、1:1.27、1:1.29、1:1.3、1:1.31、1:1.33、1:1.35、1:1.37、1:1.39、1:1.4、1:1.41、1:1.43、1:1.45、1:1.47或1:1.49等。
[0029]
优选地,步骤(1)所述3-异色满酮与亚硝酸甲酯的摩尔比为1:(1.2~1.5),例如可以为1:1.21、1:1.23、1:1.25、1:1.27、1:1.29、1:1.3、1:1.31、1:1.33、1:1.35、1:1.37、1:
1.39、1:1.4、1:1.41、1:1.43、1:1.45、1:1.47或1:1.49等。
[0030]
优选地,步骤(1)所述肟化反应在溶剂存在下进行。
[0031]
优选地,所述溶剂为醇类溶剂。
[0032]
优选地,所述醇类溶剂选自甲醇、乙醇或叔丁醇中的任意一种或至少两种的组合,进一步优选为甲醇。
[0033]
优选地,步骤(1)所述肟化反应的温度为10~35℃,例如可以为11℃、13℃、15℃、17℃、19℃、20℃、21℃、23℃、25℃、27℃、29℃、30℃、31℃、33℃或34℃,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值,进一步优选为15~20℃。
[0034]
优选地,步骤(1)所述肟化反应的时间为8~15h,例如可以为8.5h、9h、9.5h、10h、10.5h、11h、11.5h、12h、12.5h、13h、13.5h、14h或14.5h,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
[0035]
优选地,步骤(1)所述肟化反应在搅拌条件下进行。
[0036]
优选地,步骤(1)所述肟化反应在保护气氛中进行,所述保护气氛优选为氮气。
[0037]
优选地,步骤(1)所述亚硝酸甲酯的通入温度≤20℃,例如19.5℃、19℃、18.5℃、18℃、17.5℃、17℃、16.5℃、16℃、15.5℃、15℃、14.5℃、14℃、13.5℃、13℃、12.5℃、12℃、11.5℃或11℃等。
[0038]
优选地,步骤(2)所述一氯甲烷与步骤(1)所述3-异色满酮的摩尔比为(1.2~2.0):1,例如1.25:1、1.3:1、1.35:1、1.4:1、1.45:1、1.5:1、1.52:1、1.55:1、1.58:1、1.6:1、1.62:1、1.65:1、1.68:1、1.7:1、1.72:1、1.75:1、1.78:1、1.8:1、1.85:1、1.9:1或1.95:1等,进一步优选为(1.5~1.8):1。
[0039]
优选地,步骤(2)所述醚化反应在溶剂存在下进行。
[0040]
优选地,所述溶剂为非质子极性溶剂。
[0041]
优选地,所述非质子极性溶剂包括n,n-二甲基甲酰胺、n,n-二甲基乙酰胺或二甲基亚砜中的任意一种或至少两种的组合。
[0042]
优选地,步骤(2)所述一氯甲烷的通入温度为5~10℃,例如可以为5.2℃、5.5℃、5.8℃、6℃、6.2℃、6.5℃、6.8℃、7℃、7.2℃、7.5℃、7.8℃、8℃、8.2℃、8.5℃、8.8℃、9℃、9.2℃、9.5℃或9.8℃,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。通过控制一氯甲烷的通入温度,使一氯甲烷在体系中具有比较高的溶解度。
[0043]
优选地,步骤(2)所述醚化反应的温度为40~50℃,例如可以为40.5℃、41℃、41.5℃、42℃、42.5℃、43℃、43.5℃、44℃、44.5℃、45℃、45.5℃、46℃、46.5℃、47℃、47.5℃、48℃、48.5℃、49℃或49.5℃,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
[0044]
优选地,步骤(2)所述醚化反应的时间为6~12h,例如可以为6.5h、7h、7.5h、8h、8.5h、9h、9.5h、10h、10.5h、11h或11.5h,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
[0045]
为了保证醚化反应的快速进行和较高的转化率,步骤(2)所述醚化反应优选在高压釜中进行,所述醚化反应的压力为0.05~0.3mpa,例如可以为0.06mpa、0.08mpa、0.1mpa、
0.11mpa、0.13mpa、0.15mpa、0.17mpa、0.19mpa、0.2mpa、0.21mpa、0.23mpa、0.25mpa、0.27mpa或0.29mpa,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。所述压力为一氯甲烷的升温膨胀压力,随着醚化反应的进行,一氯甲烷被消耗,釜内压力逐渐下降。
[0046]
优选地,步骤(2)所述醚化反应完成后还包括产物的后处理。
[0047]
优选地,所述后处理的方法包括:将反应产物过滤、脱溶后,经复溶、水洗、干燥,得到所述3-酮-4-(甲氧亚氨基)异色满的溶液。
[0048]
优选地,所述复溶的溶剂选自甲苯、苯、二甲苯或三甲苯中的任意一种或至少两种的组合。
[0049]
优选地,所述干燥的方法包括使用干燥剂(如硫酸钠)或回流分水(温度≤80℃),通过干燥能够使3-酮-4-(甲氧亚氨基)异色满的溶液中的水分含量尽可能降低,从而减少步骤(3)中酰氯化试剂的消耗、避免副反应发生。
[0050]
优选地,步骤(3)所述酰氯化试剂为固体光气。
[0051]
优选地,所述固体光气与步骤(1)所述3-异色满酮的摩尔比为(0.6~0.8):1,例如可以为0.61:1、0.63:1、0.65:1、0.67:1、0.69:1、0.7:1、0.71:1、0.73:1、0.75:1、0.77:1或0.79:1等。
[0052]
优选地,步骤(3)所述氯化反应在催化剂存在下进行。
[0053]
优选地,所述催化剂为有机碱类化合物。
[0054]
优选地,所述有机碱类化合物选自吡啶、三乙胺或n,n-二甲基苯胺中的任意一种或至少两种的组合,进一步优选为吡啶。
[0055]
优选地,所述催化剂与酰氯化试剂的摩尔比为(0.0005~0.02):1,例如可以为0.0006:1、0.0008:1、0.001:1、0.003:1、0.005:1、0.007:1、0.009:1、0.01:1、0.011:1、0.012:1、0.013:1、0.014:1、0.015:1、0.016:1、0.017:1、0.018:1或0.019:1等。
[0056]
优选地,步骤(3)所述氯化反应在溶剂存在下进行。
[0057]
优选地,所述溶剂选自甲苯、苯、二甲苯或三甲苯中的任意一种或至少两种的组合。
[0058]
优选地,步骤(3)所述氯化反应的温度为20~60℃,例如可以为22℃、25℃、28℃、30℃、32℃、35℃、38℃、40℃、42℃、45℃、48℃、50℃、52℃、55℃或58℃,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值,进一步优选为40~50℃。
[0059]
优选地,步骤(3)所述氯化反应的时间为2~6h,例如可以为2.2h、2.5h、2.8h、3h、3.2h、3.5h、3.8h、4h、4.2h、4.5h、4.8h、5h、5.2h、5.5h或5.8h,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
[0060]
优选地,步骤(3)所述氯化反应的具体方法包括:将酰氯化试剂、催化剂与溶剂混合,得到混合液;向所述混合液中滴加3-酮-4-(甲氧亚氨基)异色满的溶液,完成滴加后40~60℃保温反应2~5h,得到所述2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酰氯。
[0061]
作为本发明的优选技术方案,所述氯化反应将3-酮-4-(甲氧亚氨基)异色满的溶液滴加入混合液中,采用“反滴加”的方式大大提高了原料转化率,使反应更加彻底,收率更高,反应产生的氯化氢可以回收制成副产盐酸。
[0062]
优选地,所述混合的温度为10~15℃,例如可以为10.5℃、11℃、11.5℃、12℃、12.5℃、13℃、13.5℃、14℃或14.5℃,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
[0063]
优选地,所述滴加的温度为40~45℃,例如可以为40.5℃、41℃、41.5℃、42℃、42.5℃、43℃、43.5℃、44℃或44.5℃,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
[0064]
优选地,步骤(3)所述氯化反应完成后还包括脱溶的步骤。
[0065]
优选地,步骤(4)所述甲醇与步骤(1)所述3-异色满酮的摩尔比为(4~6.5):1,例如可以为4.1:1、4.3:1、4.5:1、4.7:1、4.9:1、5:1、5.1:1、5.3:1、5.5:1、5.7:1、5.9:1、6:1、6.1:1、6.2:1、6.3:1或6.4:1等。
[0066]
优选地,步骤(4)所述酯化反应的温度为15~20℃,例如可以为15.5℃、16℃、16.5℃、17℃、17.5℃、18℃、18.5℃、19℃或19.5℃,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
[0067]
优选地,步骤(4)所述酯化反应的时间为2~8h,例如可以为2.5h、3h、3.5h、4h、4.5h、5h、5.5h、6h、6.5h、7h或7.5h,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
[0068]
优选地,步骤(4)所述酯化反应在负压条件下进行,能够使反应生成的氯化氢快速从反应体系中移除,有利于酯化反应的正向进行,加速反应进度,产物收率高,可以达到80%以上(以3-异色满酮计),并实现尾气氯化氢的回收。
[0069]
优选地,步骤(4)所述酯化反应的压力为-0.03~-0.01mpa,例如可以为-0.029mpa、-0.027mpa、-0.025mpa、-0.023mpa、-0.021mpa、-0.02mpa、-0.019mpa、-0.017mpa、-0.015mpa、-0.013mpa或-0.011mpa,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
[0070]
优选地,步骤(4)所述酯化反应完成后还包括产物的后处理。
[0071]
优选地,所述后处理的方法包括:将反应产物脱溶、重结晶、过滤,得到所述2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯的纯品。
[0072]
优选地,所述脱溶的方法为减压蒸馏。
[0073]
优选地,所述重结晶的溶剂为烷烃类溶剂,或烷烃类溶剂与甲醇的混合物。
[0074]
优选地,所述烷烃类溶剂为石油醚。
[0075]
优选地,所述重结晶的温度为-5~5℃,例如可以为-4.5℃、-4℃、-3.5℃、-3℃、-2.5℃、-2℃、-1.5℃、-1℃、-0.5℃、0℃、0.5℃、1℃、1.5℃、2℃、2.5℃、3℃、3.5℃、4℃或4.5℃,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
[0076]
上述重结晶步骤可以使2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯纯度(含量)达到97%以上,高含量的2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯与间三氟甲基苯乙酮肟进行缩合反应,使肟菌酯的收率更高,肟菌酯粗品的纯度(含量)高,纯化过程简单,提纯损失少,总收率更高。
[0077]
优选地,步骤(5)所述间三氟甲基苯乙酮肟与2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯的摩尔比为(1~1.2):1,例如可以为1.01:1、1.03:1、1.05:1、1.07:1、1.09:1、
1.1:1、1.11:1、1.13:1、1.15:1、1.17:1或1.19:1等。
[0078]
优选地,步骤(5)所述缩合反应在催化剂存在下进行。
[0079]
优选地,所述催化剂为金属碘盐,进一步优选为碘化钾和/或碘化钠;所述金属碘盐能够加速反应进行,克服2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯反应活性不高的缺陷,提升缩合反应的效率和选择性。
[0080]
优选地,所述催化剂与2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯的摩尔比为(0.005~0.02):1,例如可以为0.006:1、0.008:1、0.01:1、0.011:1、0.012:1、0.013:1、0.014:1、0.015:1、0.016:1、0.017:1、0.018:1或0.019:1等。
[0081]
优选地,步骤(5)所述缩合反应在缚酸剂存在下进行。
[0082]
优选地,所述缚酸剂为碳酸钾,进一步优选为粉末状碳酸钾;粉末状碳酸钾的反应速度快,反应效果最佳。
[0083]
优选地,所述粉末状碳酸钾的粒径为180~300目,例如可以为190目、200目、210目、220目、230目、240目、250目、260目、270目、280目或290目等。
[0084]
优选地,所述缚酸剂与2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯的摩尔比为(1.1~2):1,例如可以为1.12:1、1.15:1、1.2:1、1.25:1、1.3:1、1.35:1、1.4:1、1.45:1、1.5:1、1.55:1、1.6:1、1.65:1、1.7:1、1.75:1、1.8:1、1.85:1、1.9:1或1.95:1等。
[0085]
优选地,步骤(5)所述缩合反应在溶剂存在下进行。
[0086]
优选地,所述溶剂为非质子极性溶剂。
[0087]
优选地,所述非质子极性溶剂包括n,n-二甲基甲酰胺、n,n-二甲基乙酰胺或二甲基亚砜中的任意一种或至少两种的组合。
[0088]
优选地,步骤(5)所述缩合反应的温度为45~55℃,例如可以为45.5℃、46℃、46.5℃、47℃、47.5℃、48℃、48.5℃、49℃、49.5℃、50℃、51℃、52℃、53℃、54℃或54.5℃,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
[0089]
优选地,步骤(5)所述缩合反应的时间为4~8h,例如可以为4.2h、4.5h、4.8h、5h、5.2h、5.5h、5.8h、6h、6.2h、6.5h、6.8h、7h、7.2h、7.5h或7.8h,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
[0090]
优选地,步骤(5)所述缩合反应的具体方法包括:将间三氟甲基苯乙酮肟、缚酸剂、催化剂与溶剂混合,得到混合液;向所述混合液中滴加2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯,完成滴加后45~55℃保温反应3~7h,得到所述肟菌酯。
[0091]
优选地,步骤(5)所述缩合反应完成后还包括产物的后处理。
[0092]
优选地,所述后处理的方法包括:将反应产物过滤、脱溶,得到肟菌酯粗品;所述肟菌酯粗品经过重结晶、过滤和干燥,得到所述肟菌酯的纯品。
[0093]
优选地,所述重结晶的溶剂为醇类溶剂,或醇类溶剂与水的混合物,进一步优选为甲醇或甲醇的水溶液。
[0094]
优选地,所述醇类溶剂与水的混合物中醇类溶剂的浓度≥90%。
[0095]
优选地,所述重结晶的温度为0~5℃,例如可以为0.5℃、1℃、1.5℃、2℃、2.5℃、3℃、3.5℃、4℃或4.5℃,以及上述点值之间的具体点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
[0096]
优选地,所述制备方法具体包括如下步骤:
[0097]
(1)将有机金属醇盐滴加至3-异色满酮和醇类溶剂的混合物中,滴加完成后搅拌均匀,向体系中通入亚硝酸甲酯,控制体系温度≤20℃;通气结束后10~35℃反应8~10h,脱溶,得到3-异色满酮-4-亚硝酸金属盐;所述3-异色满酮、有机金属醇盐与亚硝酸甲酯的摩尔比为为1:(1.2~1.5):(1.2~1.5);
[0098]
(2)将步骤(1)得到的3-异色满酮-4-亚硝酸金属盐与非质子极性溶剂加入高压釜,在5~10℃条件下向釜中通入一氯甲烷;通气结束后关闭高压釜,升温至40~50℃醚化反应6~12h;反应完成后将产物过滤、脱溶,再经过复溶、水洗、干燥,得到所述3-酮-4-(甲氧亚氨基)异色满的溶液;所述一氯甲烷与步骤(1)所述3-异色满酮的摩尔比为(1.2~2.0):1;
[0099]
(3)将固体光气、催化剂与溶剂在10~15℃混合,得到混合液;向所述混合液中滴加步骤(2)得到的3-酮-4-(甲氧亚氨基)异色满的溶液,控制滴加温度为40~45℃;完成滴加后40~60℃保温反应2~5h,脱溶,得到2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酰氯;所述催化剂为有机碱类化合物,所述催化剂与固体光气的摩尔比为(0.0005~0.02):1,所述固体光气与步骤(1)所述3-异色满酮的摩尔比为(0.6~0.8):1;
[0100]
(4)向步骤(3)得到的2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酰氯中滴加甲醇,控制体系温度≤20℃;完成滴加后在15~20℃、-0.03~-0.01mpa条件下酯化反应2~6h;反应完成后将产物脱溶、重结晶、过滤,得到2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯;所述甲醇与步骤(1)所述3-异色满酮的摩尔比为(4~6.5):1;
[0101]
(5)将间三氟甲基苯乙酮肟、催化剂、碳酸钾与溶剂混合,得到混合液;在45~50℃条件下向所述混合液中滴加步骤(4)得到的2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯,完成滴加后45~55℃保温反应3~7h;反应完成后将产物过滤、脱溶,得到肟菌酯粗品;所述肟菌酯粗品经重结晶、过滤和干燥,得到所述肟菌酯的纯品;所述间三氟甲基苯乙酮肟、催化剂、碳酸钾与2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯的摩尔比为(1~1.2):(0.005~0.02):(1.1~2):1。
[0102]
相对于现有技术,本发明具有以下有益效果:
[0103]
本发明提供的制备方法以3-异色满酮为起始原料,经肟化反应、醚化反应、氯化反应和酯化反应,得到2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯,原子经济性好,收率高,四步反应的总收率达到85%以上,2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯的纯度≥97%,而且整个过程的中间体无需分离,三废少,适合工业化生产。将酯化反应得到的2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯与间三氟甲基苯乙酮肟进行缩合反应,得到的肟菌酯的收率≥93%,纯度达到98%以上。所述制备方法的反应条件温和,不涉及剧毒性原料,无需复杂的中间产物处理步骤,三废产生量少,能够获得高收率和高纯度的肟菌酯,适合大规模的工业化生产。
具体实施方式
[0104]
下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
[0105]
本发明以下实施例中的含量(纯度)通过高效液相色谱(hplc,lc-20at,日本岛津)
外标法测得。
[0106]
实施例1
[0107]
一种肟菌酯的制备方法,具体包括如下步骤:
[0108]
(1)向反应瓶中加入3-异色满酮(纯度99%,149.7g,1.0mol)和300g甲醇,降温至18℃,缓慢滴加甲醇钠(浓度30%,216.08g,1.2mol)的甲醇溶液,约1h滴加完毕,搅拌0.5h;向体系中通入亚硝酸甲酯气体,控制温度不超过20℃,约3h通入量达到74.0g(纯度99%,1.2mol),停止通气,20℃保温反应9h,取样分析3-异色满酮小于0.5%,减压脱溶后得到釜残,即3-异色满酮-4-亚硝酸钠213.8g,无需处理直接用于下一步;
[0109]
(2)向步骤(1)得到的3-异色满酮-4-亚硝酸钠中加入500g n,n-二甲基甲酰胺(dmf),搅拌,转移至高压釜,搅拌降温至10℃,缓慢通入一氯甲烷气体(纯度99%,76.5g,1.5mol),保持温度不高于10℃,通完一氯甲烷后,关闭高压釜;缓慢升温至40℃,保温反应8h,反应结束,降温,开釜转移物料,过滤得到3-酮-4-(甲氧亚氨基)异色满的dmf溶液,脱溶回收dmf,得到3-酮-4-(甲氧亚氨基)异色满粗品;向3-酮-4-(甲氧亚氨基)异色满粗品中加入500g甲苯和100g水,搅拌溶解,分层,用100g水洗涤两次,有机层用硫酸钠干燥,过滤,得到3-酮-4-(甲氧亚氨基)异色满的甲苯液,直接用于下一步反应;
[0110]
(3)将固体光气(纯度99%,209.8g,0.7mol)分批加入到装有500g甲苯和吡啶(0.06g,0.00076mol)的混合液中,控制温度为15℃,搅拌至固体全部溶解,得到混合液;将步骤(2)得到的3-酮-4-(甲氧亚氨基)异色满的甲苯液缓慢滴加至所述混合液中,控制温度为45℃,滴加完毕后,45℃保温反应3h,取样分析3-酮-4-(甲氧亚氨基)异色满小于0.5%,停止反应,升温脱溶,得到棕黄色粘稠状物2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酰氯;
[0111]
(4)开启微负压,使釜内压力为-0.02mpa,向步骤(3)得到的2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酰氯中缓慢加入200g甲醇,3h滴加完毕,控制温度不超过20℃,保温反应3h,反应结束后减压蒸馏回收甲醇,得到2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯粗品;向上述粗品中加入石油醚300g,搅拌后0℃结晶,过滤,得到类白色固体2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯211.6g,含量为97.3%,收率为85.2%(以3-异色满酮计);
[0112]
(5)向反应瓶中加入间三氟甲基苯乙酮肟(纯度99%,215.5g,1.05mol)、400g dmf和碳酸钾(纯度99%,209.1g,1.5mol)和催化剂碘化钾(纯度99%,1.68g,0.01mol),升温至48℃,滴加步骤(4)得到的2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯(纯度97.3%,248.4g,1.0mol,将其溶于100g dmf中配成溶液),约1h滴加完毕,45℃保温反应5h,停止反应,过滤除去盐,滤液蒸馏脱溶,得到粗品肟菌酯;向上述粗品肟菌酯中加入300g 90%的甲醇水溶液,升温溶解后降温至3℃结晶,过滤,干燥,得到白色固体肟菌酯388.2g,含量为98.2%,收率为93.5%。
[0113]
肟菌酯的结构检测:
[0114]1h-nmr(cdcl3,500mhz):δ2.17(s,3h,-n=c-ch3),3.70(s,3h,-cooch3),3.91(s,3h,n-och3),5.06(s,2h,ar-ch2on=),7.20-7.890(m,8h,arh)。
[0115]
实施例2
[0116]
一种肟菌酯的制备方法,具体包括如下步骤:
[0117]
(1)向反应瓶中加入3-异色满酮(纯度99%,149.7g,1.0mol)和300g甲醇,降温至15℃,缓慢滴加甲醇钠(浓度30%,270.1g,1.5mol)的甲醇溶液,约1.5h滴加完毕,搅拌
1.5h;向体系中通入亚硝酸甲酯气体,控制温度不超过20℃,约3h通入量达到92.5g(纯度99%,1.5mol),停止通气,15℃保温反应11h,取样分析3-异色满酮小于0.5%,减压脱溶后得到釜残,即3-异色满酮-4-亚硝酸钠214.5g,无需处理直接用于下一步;
[0118]
(2)向步骤(1)得到的3-异色满酮-4-亚硝酸钠中加入500g n,n-二甲基乙酰胺(dmac),搅拌均匀,转移至高压釜,搅拌降温至5℃,缓慢通入一氯甲烷气体(纯度99%,61.2g,1.2mol),保持温度不高于10℃,通完一氯甲烷后,关闭高压釜;缓慢升温至50℃,保温反应6h,反应结束,降温,开釜转移物料,过滤得到3-酮-4-(甲氧亚氨基)异色满的dmac溶液,脱溶回收dmac溶剂,得到3-酮-4-(甲氧亚氨基)异色满粗品;向3-酮-4-(甲氧亚氨基)异色满粗品中加入500g甲苯和100g水,搅拌溶解,分层,用100g水洗涤两次,有机层用硫酸钠干燥,过滤,得到3-酮-4-(甲氧亚氨基)异色满的甲苯液,直接用于下一步反应;
[0119]
(3)将固体光气(纯度99%,180g,0.6mol)分批加入到装有500g甲苯和吡啶(0.07g,0.0009mol)的混合液中,控制温度为10℃,搅拌至固体全部溶解,得到混合液;将步骤(2)得到的3-酮-4-(甲氧亚氨基)异色满的甲苯液缓慢滴加至所述混合液中,控制温度为40℃,滴加完毕后,40℃保温反应5h,取样分析3-酮-4-(甲氧亚氨基)异色满小于0.5%,停止反应,升温脱溶,得到棕黄色粘稠状物2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酰氯;
[0120]
(4)开启微负压,使釜内压力为-0.01mpa,向步骤(3)得到的2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酰氯中缓慢加入140g甲醇,2.5h滴加完毕,控制温度不超过20℃,保温反应2.5h,反应结束后减压蒸馏回收甲醇,得到2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯粗品;向上述粗品中加入石油醚300g,搅拌后-5℃结晶,过滤,得到类白色固体2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯214.1g,含量为97.1%,收率为86.1%(以3-异色满酮计);
[0121]
(5)向反应瓶中加入间三氟甲基苯乙酮肟(纯度99%,225.7g,1.1mol)、400g dmf和碳酸钾粉末(纯度99%,167.3g,1.2mol)和催化剂碘化钾(纯度99%,1.35g,0.008mol),升温至45℃,滴加步骤(4)得到的2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯(纯度97.1%,248.4g,1.0mol,将其溶于100g dmf中配成溶液),约1h滴加完毕,45℃保温反应7h,停止反应,过滤除去盐,滤液蒸馏脱溶,得到粗品肟菌酯;向上述粗品肟菌酯中加入300g 90%的甲醇水溶液,升温溶解后降温至0℃结晶,过滤,干燥,得到白色固体肟菌酯384.8g,含量为98.7%,收率为93.1%。
[0122]
实施例3
[0123]
一种肟菌酯的制备方法,具体包括如下步骤:
[0124]
(1)向反应瓶中加入3-异色满酮(纯度99%,149.7g,1.0mol)和300g甲醇,降温至20℃,缓慢滴加甲醇钠(浓度30%,216.1g,1.2mol)的甲醇溶液,约1h滴加完毕,搅拌0.5h;向体系中通入亚硝酸甲酯气体,控制温度不超过20℃,约3h通入量达到86.3g(纯度99%,1.4mol),停止通气,20℃保温反应8h,取样分析3-异色满酮小于0.5%,减压脱溶后得到釜残,即3-异色满酮-4-亚硝酸钠212.7g,无需处理直接用于下一步;
[0125]
(2)向步骤(1)得到的3-异色满酮-4-亚硝酸钠中加入500g dmf,搅拌均匀,转移至高压釜,搅拌降温至10℃,缓慢通入一氯甲烷气体(纯度99%,91.8g,1.8mol),保持温度不高于10℃,通完一氯甲烷后,关闭高压釜;缓慢升温至40℃,保温反应12h,反应结束,降温,开釜转移物料,过滤得到3-酮-4-(甲氧亚氨基)异色满的dmf溶液,脱溶回收dmf溶剂,得到
3-酮-4-(甲氧亚氨基)异色满粗品;向3-酮-4-(甲氧亚氨基)异色满粗品中加入500g甲苯和100g水,搅拌溶解,分层,用100g水洗涤两次,有机层用硫酸钠干燥,过滤,得到3-酮-4-(甲氧亚氨基)异色满的甲苯液,直接用于下一步反应;
[0126]
(3)将固体光气(纯度99%,239.8g,0.8mol)分批加入到装有500g甲苯和吡啶(0.095g,0.0012mol)的混合液中,控制温度为15℃,搅拌至固体全部溶解,得到混合液;将步骤(2)得到的3-酮-4-(甲氧亚氨基)异色满的甲苯液缓慢滴加至所述混合液中,控制温度为45℃,滴加完毕后,60℃保温反应2.5h,取样分析3-酮-4-(甲氧亚氨基)异色满小于0.5%,停止反应,升温脱溶,得到棕黄色粘稠状物2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酰氯;
[0127]
(4)开启微负压,使釜内压力为-0.03mpa,向步骤(3)得到的2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酰氯中缓慢加入175g甲醇,2.5h滴加完毕,控制温度不超过20℃,保温反应4h,反应结束后减压蒸馏回收甲醇,得到2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯粗品;向上述粗品中加入石油醚300g,搅拌后-5℃结晶,过滤,得到类白色固体2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯213.5g,含量为97.2%,收率为85.8%(以3-异色满酮计);
[0128]
(5)向反应瓶中加入间三氟甲基苯乙酮肟(纯度99%,246.2g,1.2mol)、400g dmf和碳酸钾粉末(纯度99%,251g,1.8mol)和催化剂碘化钾(纯度99%,3.36g,0.02mol),升温至50℃,滴加步骤(4)得到的2-(2-(氯甲基)苯基)-2-(甲氧亚胺基)乙酸甲酯(纯度97.2%,248.5g,1.0mol,将其溶于100g dmf中配成溶液),约1h滴加完毕,55℃保温反应4h,停止反应,过滤除去盐,滤液蒸馏脱溶,得到粗品肟菌酯;向上述粗品肟菌酯中加入300g 90%的甲醇水溶液,升温溶解后降温至5℃结晶,过滤,干燥,得到白色固体肟菌酯389.1g,含量为98.1%,收率为93.6%。
[0129]
申请人声明,本发明通过上述实施例来说明本发明的一种肟菌酯的制备方法,但本发明并不局限于上述实施例,即不意味着本发明必须依赖上述实施例才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1