杂环亚基乙酰胺衍生物的制造方法与流程
1.本发明涉及作为杂环亚基乙酰胺衍生物(heterocyclidene acetamide derivative)的下述式(i)的(e)-2-(7-三氟甲基色满-4-亚基)-n-((7r)-7-羟基-5,6,7,8-四氢萘-1-基)乙酰胺的新型制造方法。另外,本发明涉及对下述式(i)的化合物的制造有用的中间体即下述式(b)的(r)-8-氨基-1,2,3,4-四氢萘-2-醇或其盐的新型制造方法。
背景技术:
2.式(i)的(e)-2-(7-三氟甲基色满-4-亚基)-n-((7r)-7-羟基-5,6,7,8-四氢萘-1-基)乙酰胺是trpv1拮抗剂(trpv1=transient receptor potential vanilloid 1),被期待作为trpv1受体所参与的疾病(例如,疼痛(例如,神经性疼痛、糖尿病性神经痛、手术后疼痛、骨关节炎、类风湿性关节炎痛、炎症性疼痛、癌性疼痛、偏头痛等)、神经障碍、神经损伤、神经变性、慢性阻塞性肺疾病、哮喘、鼻炎、眼等的粘膜炎症、神经性皮肤疾病、炎症性皮肤疾病、过敏性疾病、尿失禁、急迫性尿失禁、过活动性膀胱、膀胱炎、或瘙痒症等)的预防和/或治疗剂(专利文献1)。
3.[化学式1]式(i)的化合物的制造方法公开在国际公开第2007/010383号小册子(专利文献1)中。在该文献中,式(i)的化合物通过下述(图解a)中所示的《工序1》~《工序3》的工序进行制造。
[0004]
《工序1》 通过使用依据文献已知的方法(例如,国际公开第2005/040100号小册子(专利文献2)等)而制造的8-氨基-3,4-二氢萘-2(1h)-酮(式(im-3))、(e)-2-(7-(三氟甲基)色满-4-亚基)乙酸(式(ca-1) [cas no. 920334-15-2]、非专利文献1)和缩合剂(1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐(edci
・
hcl))进行缩合反应,得到式(im-k)的化合物。
[0005]
《工序2》 通过将式(im-k)的化合物用硼氢化钠进行还原,得到式(i-rac)的化合物。
[0006]
《工序3》 通过将式(i-rac)的化合物利用光学活性柱进行光学拆分,得到作为式(i)的化合物及其异构体的式(i-s)的化合物。
[0007]
然而,该制造方法在最终工序中进行柱拆分,得到式(i)的化合物,难以再利用在柱拆分后得到的式(i-s)的化合物。
[0008]
[化学式2](图解a)
另一方面,相当于式(i)的部分结构式的下述式(b):[化学式3]的化合物的制造方法公开在国际公开第2005/040100号小册子(专利文献2)、国际公开第2003/095420号小册子(专利文献3)、国际公开第2005/040119号小册子(专利文献4)和国际公开第2010/127855号小册子(专利文献5)中。在该文献中,将以8-氨基萘-2-醇(式(sm-1))作为起始物质经过酚基的烷基化、birch还原、烷基的脱保护反应而得到的8-氨基-3,4-二氢萘-2(1h)-酮(式(im-3))在ru催化剂存在下进行不对称还原,由此制造式(b)的化合物(图解1)。
[0009]
然而,该制造方法需要在其工序中使用birch还原和在最终工序的不对称还原中使用金属(ru)催化剂以降低所得化合物中的金属(ru)的残留率的工序。
[0010]
[化学式4](图解1)此外,式(b)的化合物的制造方法还公开在国际公开第2009/050289号小册子(专利文献6)、国际公开第2010/045401号小册子(专利文献7)和国际公开第2010/045402号小
册子(专利文献8)中。在该文献中,以8-氨基萘-2-醇(式(sm-1))作为起始物质,通过环选择性地还原萘环而衍生成作为外消旋体的8-氨基-1,2,3,4-四氢萘-2-醇(式a),之后使用光学活性柱进行拆分,由此制造式(b)的化合物(图解2)。
[0011]
然而,该制造方法难以再利用在柱拆分后得到的其它异构体(s型)。
[0012]
[化学式5](图解2)而且,在国际公开第2009/055749号小册子(专利文献9)中还公开了式(b)的化合物的制造方法。在该文献中,通过在式(a)的外消旋体中导入手性辅助基团,将在进行非对映异构拆分后得到的非对映异构体利用柱进行拆分,由此制造式(b)的化合物(图解3)。
[0013]
然而,该制造方法也难以再利用在柱拆分后得到的其他异构体(s型)。
[0014]
[化学式6](图解3)上述各文献中公开的制造式(b)的化合物的方法因制造过程的反应种类、使用的试剂、利用柱拆分外消旋体或者非对映异构体,而具有拆分后的其它异构体难以再利用等的课题,在式(b)的化合物的大量合成或工业生产中需求它的改良制造方法。即,在考虑式(b)的化合物的大量合成或工业生产的情况下,需求找到不同于上述各文献记载的制造方法的新型制造方法。由于以高收率且高光学纯度大量合成式(b)的化合物的制造方法尚属未知,所以认为如果能够找到以短工序、高化学收率、且高光学纯度大量合成式(b)的化合物的制造方法,则可解决制造式(b)的化合物的上述课题。
[0015]
以tempo (2,2,6,6-四甲基哌啶-1-氧自由基)作为氧化剂将仲醇氧化成酮的方法公开在美国专利第5136103号说明书(专利文献10)等中。然而,将在分子内具有取代氨基和羟基的1,2,3,4-四氢萘(例如,叔丁基-(7-羟基-5,6,7,8-四氢萘-1-基)氨基甲酸酯等)用于原料的、tempo氧化反应是未知的。另外,将该化合物用于原料的、基于流动化学(流动反应)的tempo氧化反应也是未知的。
[0016]
在美国专利第5225339号说明书(专利文献11)或美国专利第5342767号说明书(专利文献12)中公开了利用来自克菲尔乳杆菌(lactobacillus kefir)的还原酶进行的酮的还原,但适应于保护基取代氨基-3,4-二氢萘-2(1h)-酮或者β-四氢萘酮这样的酮化合物的酶还原还没有公开。
[0017]
在advanced synthesis&catalysis、350 (14+15)、第2322-2328页、2008年(非专利文献2)中公开了α-四氢萘酮或β-四氢萘酮的酮的酶还原(还原酶:来自克菲尔乳酸菌)。然而,已明确指出:在使用来自克菲尔乳酸菌的还原酶的情况下,β-四氢萘酮的酮基的还原不进行。
[0018]
在国际公开第2018/205948号小册子(专利文献13)中公开了8-溴-1,2,3,4-四氢萘-2-醇([cas no.444619-84-5]、非专利文献3)及其制造方法,但作为其手性体之一的(r)-8-溴-1,2,3,4-四氢萘-2-醇及其制造方法是未知的。
[0019]
在cas registry中公开了8-氟-1,2,3,4-四氢萘-2-醇([cas no.1823867-35-1]、非专利文献4),但作为其手性体之一的(r)-8-氟-1,2,3,4-四氢萘-2-醇及其制造方法是未知的。另外,在cas registry中公开了8-氯-1,2,3,4-四氢萘-2-醇([cas no.1823929-47-0]、非专利文献5),但作为其手性体之一的(r)-8-氯-1,2,3,4-四氢萘-2-醇及其制造方法是未知的。
[0020]
在bioorganic & medicinal chemistry letters、18(6)、第1830-1834页、2008年(非专利文献6)中公开了:通过对8-溴-6-氟-1,2,3,4-四氢萘-2-醇进行使用pd催化剂(pd2(dba)3)和氨基甲酸叔丁酯的氨基化反应、接着进行boc基团的脱保护,来制造8-氨基-6-氟-1,2,3,4-四氢萘-2-醇的方法(收率18%)。
[0021]
现有技术文献专利文献专利文献1:国际公开第2007/010383号小册子;专利文献2:国际公开第2005/040100号小册子;专利文献3:国际公开第2003/095420号小册子;专利文献4:国际公开第2005/040119号小册子;专利文献5:国际公开第2010/127855号小册子;专利文献6:国际公开第2009/050289号小册子;专利文献7:国际公开第2010/045401号小册子;专利文献8:国际公开第2010/045402号小册子;专利文献9:国际公开第2009/055749号小册子;专利文献10:美国专利第5136103号说明书;专利文献11:美国专利第5225339号说明书;专利文献12:美国专利第5342767号说明书;专利文献13:国际公开第2018/205948号小册子;非专利文献非专利文献1:cas no. 920334-15-2;非专利文献2:advanced synthesis&catalysis, 350 (14+15), 第2322-2328页, 2008年;
非专利文献3:cas no. 444619-84-5;非专利文献4:cas no. 1823867-35-1;非专利文献5:cas no. 1823929-47-0;非专利文献6:bioorganic & medicinal chemistry letters, 18(6), 第1830-1834页, 2008年。
技术实现要素:
[0022]
发明所要解决的课题基于这种状况,需求上述式(i)的化合物的新的制造方法。
[0023]
用于解决课题的手段为了解决上述课题,本发明人反复进行了深入研究。其结果,发现了收率良好、且容易地制造上述式(i)的化合物的新型的制造方法等,且根据该见解完成了本发明。即,发现了使用dmt-mm (4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉盐酸盐,(4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride) (cas no:3945-69-5)作为缩合剂,通过式(ca-1)所表示的羧酸和式(b)所表示的氨基醇或其盐的缩合反应,制造式(i)的化合物的新型方法(图解b)。
[0024]
[化学式7](图解b)另外,还发现了对制造式(i)的化合物有用的中间体即式(b)的化合物或其盐的新型的制造方法等,根据该见解完成了本发明(图解c-1)。
[0025]
[化学式8](图解c-1)
而且,还发现了对制造式(i)的化合物有用的中间体即式(b)的化合物或其盐的新型的制造方法等,根据该见解完成了本发明(图解c-2)。
[0026]
[化学式9](图解c-2)发明效果根据本发明,提供上述式(i)的化合物、或上述式(b)的化合物或其盐的新的制造方法。优选提供适合于上述式(i)的化合物、或式(b)的化合物或其盐的大量合成或工业生产的有效的制造方法。本发明的几个方案的制造方法是可收率良好、且工业上有利地制造式(i)的化合物、或式(b)的化合物或其盐的方法,产业实用性高。另外,另外几个方案提供成为用于得到式(b)所表示的化合物及其盐的原料的、式(a-7)和式(a8)所表示的新型化合物。
附图说明
[0027]
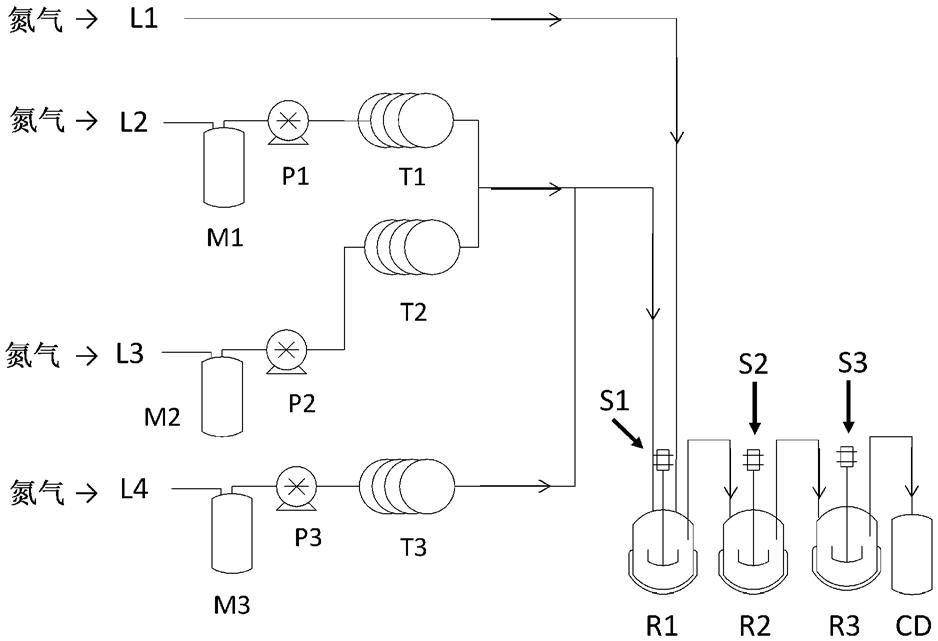
[图1]是流动化学中使用的反应装置的例子。
[0028]
[图2]是显示式(b)的化合物的hbr盐的晶体结构的图。
[0029]
[图3]是显示式(i)的化合物的晶体结构的图。
具体实施方式
[0030]
[本发明的方案]这里,提供上述式(i)的化合物的制造方法。几个方案均为以式(a)的化合物作为
起始物质的、式(i)的化合物的制造方法。另几个方案均为以式(a-5)的化合物作为起始物质的、式(i)的化合物的制造方法。另外几个方案均为以式(a-6)的化合物作为起始物质的、式(i)的化合物的制造方法。另外几个方案均为以式(a-7)的化合物作为起始物质的、式(i)的化合物的制造方法。
[0031]
另外几个方案均为以式(sm8)的化合物作为起始物质的、式(i)的化合物的制造方法。另外几个方案均为以式(sm8-br)的化合物作为起始物质的、式(i)的化合物的制造方法。另外几个方案均为以式(a8)的化合物作为起始物质的、式(i)的化合物的制造方法。另外几个方案均为以式(a8-br)的化合物作为起始物质的、式(i)的化合物的制造方法。另外几个方案均为以式(b)的化合物作为起始物质的、式(i)的化合物的制造方法。
[0032]
另外,还提供上述式(b)的化合物或其盐的制造方法。几个方案均为以式(a)的化合物作为起始物质的、式(b)的化合物或其盐的制造方法。另几个方案均为以式(a-5)的化合物作为起始物质的、式(b)的化合物或其盐的制造方法。另外几个方案均为以式(a-6)的化合物作为起始物质的、式(b)的化合物或其盐的制造方法。另外几个方案均为以式(a-7)的化合物作为起始物质的、式(b)的化合物或其盐的制造方法。
[0033]
另外几个方案均为以式(sm8)所表示的化合物作为起始物质的、式(b)所表示的化合物及其盐的制造方法。另外几个方案均为以式(sm8-br)所表示的化合物作为起始物质的、式(b)所表示的化合物及其盐的制造方法。另外几个方案均为以式(a8)所表示的化合物作为起始物质的、式(b)所表示的化合物及其盐的制造方法。另外几个方案均为以式(a8-br)所表示的化合物作为起始物质的、式(b)所表示的化合物及其盐的制造方法。
[0034]
而且,另外几个方案均为以式(a-5)或式(a-7)的化合物作为起始物质的、式(a-6)的化合物的制造方法。而且,另外几个方案均为式(a-6)的化合物作为起始物质的、式(a-7)的化合物的制造方法。而且,另外几个方案均为式(a-7)所表示的化合物。
[0035]
而且,另外几个方案均为以式(sm8)所表示的化合物作为起始物质的、式(a8)所表示的化合物的制造方法。而且,另外几个方案均为以式(sm8-br)所表示的化合物作为起始物质的、式(a8-br)所表示的化合物的制造方法。而且,另外几个方案均为上述式(a8)所表示的化合物和式(a8-br)所表示的化合物。
[0036]
以下,对各方案进行具体地记载。
[0037]
[1] 第1方案是制造式(b)所表示的化合物或其盐的方法,[化学式10]该制造方法包括下述工序:将式(a)所表示的化合物的氨基进行叔丁氧羰基化,得到式(a-5)所表示的化合物的工序,[化学式11]
[化学式12];使式(a-5)所表示的化合物进行氧化反应,得到式(a-6)所表示的化合物的工序,[化学式13];使式(a-6)所表示的化合物进行不对称还原,得到式(a-7)所表示的化合物的工序,[化学式14];以及使式(a-7)所表示的化合物的叔丁氧羰基进行脱保护,得到式(b)所表示的化合物或其盐的工序。
[0038]
[2] 第2方案是制造式(b)所表示的化合物或其盐的方法,[化学式15]该制造方法包括以下工序:使式(a-5)所表示的化合物进行氧化反应,得到式(a-6)所表示
化合物的工序,[化学式16][化学式17];使式(a-6)所表示的化合物进行不对称还原,得到式(a-7)所表示的化合物的工序,[化学式18];以及使式(a-7)所表示的化合物的叔丁氧羰基进行脱保护,得到式(b)所表示的化合物或其盐的工序。
[0039]
[3] 第3方案是制造式(b)所表示的化合物或其盐的方法,[化学式19]该制造方法包括以下工序:使式(a-6)所表示的化合物进行不对称还原,得到式(a-7)所表示的化合物的工序,[化学式20]
[化学式21];以及使式(a-7)所表示的化合物的叔丁氧羰基进行脱保护,得到式(b)所表示的化合物或其盐的工序。
[0040]
[4] 第4方案是制造式(b)所表示的化合物或其盐的方法,[化学式22]该制造方法包括下述工序:使式(a-7)所表示的化合物的叔丁氧羰基进行脱保护,得到式(b)所表示的化合物或其盐的工序,[化学式23]。
[0041]
[4b] 第4b方案是制造式(b)所表示的化合物的盐的方法,[化学式24]该制造方法包括下述工序:通过在式(b)所表示的化合物中添加酸,得到式(b)所
表示的化合物的盐的工序。
[0042]
[4b-1] 上述方案[4b]中,作为在得到式(b)所表示的化合物的盐的工序中所用的酸,优选为无机酸或有机酸,更优选为无机酸,进一步优选为氢溴酸。
[0043]
[5] 第5方案是制造式(a-6)所表示的化合物的方法,[化学式25]该制造方法包括以下工序:使式(a-5)所表示的化合物进行氧化反应,得到式(a-6)所表示的化合物的工序,[化学式26]。
[0044]
[6] 第6方案是制造式(a-7)所表示的化合物的方法,[化学式27]该制造方法包括以下工序:使式(a-6)所表示的化合物进行不对称还原,得到式(a-7)所表示的化合物的工序,[化学式28]。
[0045]
[7] 第7方案是制造式(i)所表示的化合物的方法,[化学式29]
该制造方法包括以下工序:使用dmt-mm作为缩合剂,将式(b)所表示的化合物或其盐与式(ca-1)所表示的化合物进行缩合反应,得到式(i)所表示的化合物的工序,[化学式30][化学式31]。
[0046]
[8] 第8方案是制造式(i)所表示的化合物的方法,[化学式32]该制造方法包括以下工序:将式(a)所表示的化合物的氨基进行叔丁氧羰基化,得到式(a-5)所表示的化合物的工序,[化学式33][化学式34]
;使式(a-5)所表示的化合物进行氧化反应,得到式(a-6)所表示的化合物的工序,[化学式35];使式(a-6)所表示的化合物进行不对称还原,得到式(a-7)所表示的化合物的工序,[化学式36];使式(a-7)所表示的化合物的叔丁氧羰基进行脱保护,得到式(b)所表示的化合物或其盐的工序,[化学式37];以及使用dmt-mm作为缩合剂,将式(b)所表示的化合物或其盐与式(ca-1)所表示的化合物进行缩合反应,得到式(i)所表示的化合物的工序,[化学式38]。
[0047]
[9] 第9方案是制造式(i)所表示的化合物的方法,[化学式39]
该制造方法包括以下工序:使式(a-5)所表示的化合物进行氧化反应,得到式(a-6)所表示的化合物的工序,[化学式40][化学式41];使式(a-6)所表示的化合物进行不对称还原,得到式(a-7)所表示的化合物的工序,[化学式42];使式(a-7)所表示的化合物的叔丁氧羰基进行脱保护,得到式(b)所表示的化合物或其盐的工序,[化学式43];以及使用dmt-mm作为缩合剂,将式(b)所表示的化合物或其盐与式(ca-1)所表示的化
合物进行缩合反应,得到式(i)所表示的化合物的工序,[化学式44]。
[0048]
[10] 第10方案是制造式(i)所表示的化合物的方法,[化学式45]该制造方法包括以下工序:使式(a-6)所表示的化合物进行不对称还原,得到式(a-7)所表示的化合物的工序,[化学式46][化学式47];使式(a-7)所表示的化合物的叔丁氧羰基进行脱保护,得到式(b)所表示的化合物或其盐的工序,[化学式48];以及使用dmt-mm作为缩合剂,将式(b)所表示的化合物或其盐与式(ca-1)所表示的化
合物进行缩合反应,得到式(i)所表示的化合物的工序,[化学式49]。
[0049]
[11] 第11方案是制造式(i)所表示的化合物的方法,[化学式50]该制造方法包括以下工序:使式(a-7)所表示的化合物的叔丁氧羰基进行脱保护,得到式(b)所表示的化合物或其盐的工序,[化学式51][化学式52];以及使用dmt-mm作为缩合剂,将式(b)所表示的化合物或其盐与式(ca-1)所表示的化合物进行缩合反应,得到式(i)所表示的化合物的工序,[化学式53]。
[0050]
[12] 第12方案是式(a-7)所表示的化合物,[化学式54]
。
[0051]
上述各方案中,“使式(a-7)所表示的化合物的叔丁氧羰基进行脱保护,得到式(b)所表示的化合物或其盐的工序”可进一步包括下述工序:通过将使式(a-7)所表示的化合物的叔丁氧羰基进行脱保护而得到的式(b)所表示的化合物的盐进行脱盐,得到式(b)所表示的化合物;或通过将使式(a-7)所表示的化合物的叔丁氧羰基进行脱保护而得到的式(b)所表示的化合物转化成其盐,得到式(b)所表示的化合物的盐。
[0052]
[13] 第13方案是制造式(b)所表示的化合物的方法,[化学式55]该制造方法包括以下工序:通过使式(sm8)所表示的化合物的酮基进行不对称还原,得到式(a8)所表示的化合物的工序,[化学式56][式(sm8)中,x为卤素原子][化学式57][式(a8)中,x为卤素原子];以及通过在催化剂存在下使氨水与式(a8)所表示的化合物进行反应,得到式(b)所表示的化合物的工序。
[0053]
[13-1] 上述方案[13]中,式(sm8)和式(a8)所表示的化合物中的x优选为氟原子、氯原子或溴原子,更优选为溴原子。
[0054]
[13-2] 上述方案[13]中,催化剂优选为pd催化剂或cu催化剂,更优选为cu催化剂,进一步优选为cu2o。
[0055]
[13-3] 第13-3方案是上述方案[13]的式(b)所表示的化合物的盐的制造方法,是通过在式(b)所表示的化合物中添加无机酸或有机酸而得到式(b)所表示的化合物的盐的制造方法。关于在得到式(b)所表示的化合物的盐时所用的无机酸或有机酸,优选为无机酸,更优选为盐酸或氢溴酸,进一步优选为氢溴酸。
[0056]
本说明书中,只要没有特别说明,则“卤素原子”是指,例如氟原子、氯原子、溴原子和碘原子等。
[0057]
[14] 第14方案是制造式(b)所表示的化合物的方法,[化学式58]该制造方法包括:通过在催化剂存在下使氨水与式(a8)所表示的化合物进行反应,得到式(b)所表示的化合物,[化学式59][式(a8)中,x为卤素原子]。
[0058]
[14-1] 上述方案[14]中,式(a8)所表示的化合物中的x优选为氟原子、氯原子或溴原子,更优选为溴原子。
[0059]
[14-2] 上述方案[14]中,催化剂优选为pd催化剂或cu催化剂,更优选为cu催化剂,进一步优选为cu2o。
[0060]
[14-3] 第14-3方案是上述方案[14]的式(b)所表示的化合物的盐的制造方法,是通过在式(b)所表示的化合物中添加无机酸或有机酸而得到式(b)所表示的化合物的盐的制造方法。关于在得到式(b)所表示的化合物的盐时所用的无机酸或有机酸,优选为无机酸,更优选为盐酸或氢溴酸,进一步优选为氢溴酸。
[0061]
[15] 第15方案是制造式(a8)所表示的化合物的方法,[化学式60]
[式(a8)中,x为卤素原子]该制造方法包括:通过使式(sm8)所表示的化合物的酮基进行不对称还原,得到式(a8)所表示的化合物,[化学式61][式(sm8)中,x为卤素原子]。
[0062]
[15-1] 上述方案[15]中,式(sm8)和式(a8)所表示的化合物中的x优选为氟原子、氯原子或溴原子,更优选为溴原子。
[0063]
[16] 第16方案是式(a8)所表示的化合物,[化学式62][式(a8)中,x为卤素原子]。
[0064]
[16-1] 上述方案[16]中,式(a8)所表示的化合物中的x优选为氟原子、氯原子或溴原子,更优选为溴原子。
[0065]
[17] 第17方案是制造式(i)所表示的化合物的方法,[化学式63]该制造方法包括以下工序:通过使式(sm8)所表示的化合物的酮基进行不对称还原,得到式(a8)所表示的化
合物的工序,[化学式64][式(sm8)中,x为卤素原子][化学式65][式(a8)中,x为卤素原子];在催化剂存在下使氨水与式(a8)所表示的化合物进行反应,得到式(b)所表示的化合物的工序,[化学式66];以及使用dmt-mm作为缩合剂,将式(b)所表示的化合物与式(ca-1)所表示的化合物进行缩合反应,得到式(i)所表示的化合物的工序,[化学式67]。
[0066]
[17-1] 上述方案[17]中,式(sm8)和式(a8)所表示的化合物中的x优选为氟原子、氯原子或溴原子,更优选为溴原子。
[0067]
[17-2] 上述方案[17]中,催化剂优选为pd催化剂或cu催化剂,更优选为cu催化剂,进一步优选为cu2o。
[0068]
[18] 第18方案是制造式(i)所表示的化合物的方法,[化学式68]
该制造方法包括以下工序:通过使式(sm8)所表示的化合物的酮基进行不对称还原,得到式(a8)所表示的化合物的工序,[化学式69][式(sm8)中,x为卤素原子][化学式70][式(a8)中,x为卤素原子];在催化剂存在下使氨水与式(a8)所表示的化合物进行反应,得到式(b)所表示的化合物的工序,[化学式71];通过在式(b)所表示的化合物中添加酸,得到式(b)所表示的化合物的盐的工序;以及使用dmt-mm作为缩合剂,将式(b)所表示的化合物的盐与式(ca-1)所表示的化合物进行缩合反应,得到式(i)所表示的化合物的工序,[化学式72]
。
[0069]
[18-1] 上述方案[18]中,式(sm8)和式(a8)所表示的化合物中的x优选为氟原子、氯原子或溴原子,更优选为溴原子。
[0070]
[18-2] 上述方案[18]中,催化剂优选为pd催化剂或cu催化剂,更优选为cu催化剂,进一步优选为cu2o。
[0071]
[18-3] 上述方案[18]中,作为在得到式(b)所表示的化合物的盐的工序中所用的酸,优选为无机酸或有机酸,更优选为无机酸,进一步优选为盐酸或氢溴酸,特别优选为氢溴酸。
[0072]
[19] 第19方案是制造式(i)所表示的化合物的方法,[化学式73]该制造方法包括以下工序:通过在催化剂存在下使氨水与式(a8)所表示的化合物进行反应,得到式(b)所表示的化合物工序,[化学式74][式(a8)中,x为卤素原子][化学式75];以及使用dmt-mm作为缩合剂,将式(b)所表示的化合物与式(ca-1)所表示的化合物进行缩合反应,得到式(i)所表示的化合物的工序,[化学式76]
。
[0073]
[19-1] 上述方案[19]中,式(sm8)和式(a8)所表示的化合物中的x优选为氟原子、氯原子或溴原子,更优选为溴原子。
[0074]
[19-2] 上述方案[19]中,催化剂优选为pd催化剂或cu催化剂,更优选为cu催化剂,进一步优选为cu2o。
[0075]
[20] 第20方案是制造式(i)所表示的化合物的方法,[化学式77]该制造方法包括以下工序:通过在催化剂存在下使氨水与式(a8)所表示的化合物进行反应,得到式(b)所表示的化合物的工序,[化学式78][式(a8)中,x为卤素原子][化学式79];通过在式(b)所表示的化合物中添加酸,得到式(b)所表示的化合物的盐的工序;以及使用dmt-mm作为缩合剂,将式(b)所表示的化合物的盐与式(ca-1)所表示的化合物进行缩合反应,得到式(i)所表示的化合物的工序,[化学式80]
。
[0076]
[20-1] 上述方案[20]中,式(sm8)和式(a8)所表示的化合物中的x优选为氟原子、氯原子或溴原子,更优选为溴原子。
[0077]
[20-2] 上述方案[20]中,催化剂优选为pd催化剂或cu催化剂,更优选为cu催化剂,进一步优选为cu2o。
[0078]
[20-3] 上述方案[20]中,作为在得到式(b)所表示的化合物的盐的工序中所用的酸,优选为无机酸或有机酸,更优选为无机酸,进一步优选为盐酸或氢溴酸,特别优选为氢溴酸。
[0079]
以下,针对上述方案中的各反应进行详细的说明。
[0080]
《制造式(a-5)的化合物的工序》式(a-5)的化合物是通过将式(a)的化合物的氨基进行叔丁氧羰基化而得到。
[0081]
作为叔丁氧羰基化剂,例如可举出:二碳酸二叔丁酯(boc2o)、2-(叔丁氧羰氧基亚氨基)-2-苯基乙腈(boc-on)、n-叔丁氧羰基咪唑、2-(叔丁氧羰基硫代)-4,6-二甲基嘧啶、1-叔丁氧羰基-1,2,4-三唑、碳酸叔丁基苯酯、肼基甲酸叔丁酯和n-(叔丁氧羰氧基)邻苯二甲酰亚胺等。优选为二碳酸二叔丁酯(boc2o)、2-(叔丁氧羰氧基亚氨基)-2-苯基乙腈(boc-on),更优选为二碳酸二叔丁酯(boc2o)。
[0082]
相对于1摩尔当量的式(a)的化合物,叔丁氧羰基化剂的使用量通常为1.0~2.0摩尔当量,优选为1.1~1.8摩尔当量,更优选为1.3~1.65摩尔当量。
[0083]
反应可在溶剂的存在下进行。作为溶剂,例如可使用:二氯甲烷、乙腈、二乙醚、四氢呋喃、1,2-二甲氧基乙烷、1,4-二噁烷、叔丁醚、甲苯、水等不参与反应的溶剂或它们的混合溶剂,可根据所用的叔丁氧羰基化剂的种类适当选择。优选为四氢呋喃、1,4-二噁烷、四氢呋喃-水的混合溶剂和1,4-二噁烷-水的混合溶剂,更优选为四氢呋喃、四氢呋喃-水的混合溶剂和1,4-二噁烷-水的混合溶剂。
[0084]
反应可在碱的存在下进行。作为碱,可使用碳酸氢钠、碳酸钾、碳酸钠、三乙胺、n,n-二异丙基乙胺、吡啶等碱,可根据所用的叔丁氧羰基化剂的种类适当选择。优选为碳酸氢钠、三乙胺和吡啶,更优选为碳酸氢钠。
[0085]
相对于1摩尔当量的式(a)的化合物,碱的使用量例如为1.0~4.0摩尔当量,优选为1.0~3.5摩尔当量,更优选为1.0~3.2摩尔当量。
[0086]
关于反应温度,例如可在-78℃~溶剂回流的温度的范围、-78℃~室温的范围、0℃~溶剂回流的温度的范围或0℃~室温的范围等下进行反应,可根据所用的叔丁氧羰基化剂的种类适当选择。优选为20℃~55℃的范围。
[0087]
《制造式(a-6)的化合物的工序》式(a-6)的化合物是通过使式(a-5)的化合物或式(a-7)的化合物进行氧化反应而得到。
[0088]
作为氧化反应,例如可举出:swern氧化、pcc氧化(铬酸盐氧化)、dess-martin氧
化、tpap氧化、tempo氧化等。优选为tempo氧化。
[0089]
tempo氧化通常是指,将tempo与再氧化剂组合作为氧化剂来氧化醇等底物的反应。另外,tempo氧化也可在碱的存在下进行。
[0090]
在氧化反应中,例如采用分批法和流动化学(基于使用连续搅拌槽反应器(cstr:continuous stirred tank reactor)的流动模式的反应)。
[0091]
相对于1摩尔当量的式(a-5)的化合物或式(a-7)的化合物,氧化反应中的氧化剂的使用量通常为1.0~2.2摩尔当量,优选为1.2~2.1摩尔当量,更优选为1.4~2.0摩尔当量。
[0092]
相对于1摩尔当量的式(a-5)的化合物或式(a-7)的化合物,tempo氧化中的tempo的使用量通常为0.01~1.0摩尔当量,优选为0.05~0.7摩尔当量,更优选为0.5摩尔当量。
[0093]
在tempo氧化中的再氧化剂的例子中包含次氯酸钠(naclo)或碘苯二乙酸盐等。相对于1摩尔当量的式(a-5)的化合物或式(a-7)的化合物,tempo氧化中的次氯酸钠的使用量通常为1.0~2.5摩尔当量,优选为1.1~2.2摩尔当量,更优选为1.2~2.0摩尔当量。
[0094]
tempo氧化可在碱的存在下进行,例如,相对于1摩尔当量的式(a-5)的化合物或式(a-7)的化合物,作为碱的nahco3的使用量通常为1.0~5.0摩尔当量,优选为2.0~4.5摩尔当量,更优选为4.0摩尔当量。
[0095]
相对于1摩尔当量的式(a-5)的化合物或式(a-7)的化合物,tempo氧化中的kbr的使用量通常为0.01~0.30摩尔当量,优选为0.02~0.25摩尔当量,更优选为0.05~0.2摩尔当量。
[0096]
氧化反应(例如,tempo氧化)可在溶剂的存在下进行。作为溶剂,例如可使用:二氯甲烷、1,2-二氯乙烷、氯仿、乙腈、丙酮、水等不参与反应的溶剂或它们的混合溶剂,可根据所用的氧化反应的种类适当选择。在tempo氧化中,优选为二氯甲烷、乙腈、丙酮、水或它们的混合溶剂,更优选为二氯甲烷、乙腈、丙酮、水、二氯甲烷-水、乙腈-水或丙酮-水,进一步优选为二氯甲烷、水或二氯甲烷-水。
[0097]
关于氧化反应(例如,tempo氧化的反应温度),例如可在-78℃~溶剂回流的温度的范围、-78℃~室温的范围、0℃~溶剂回流的温度的范围或0℃~室温的范围等下进行反应,可根据所用的氧化反应适当选择。在tempo氧化中,优选为-2℃~5℃的范围。
[0098]
在tempo氧化反应后,为了去除tempo,还可使用na2s2o
4 (水溶液)等还原剂。
[0099]
《制造式(a-7)的化合物的工序》式(a-7)的化合物是通过使式(a-6)的酮化合物进行不对称还原而得到。
[0100]
作为不对称还原,例如可举出:使用化学催化剂等的不对称还原、使用了生物催化剂(酵母、真菌、霉菌、酶等)的不对称还原等。优选为使用了酶的不对称还原,更优选为使用了酮还原酶((kred:keto reductase))作为酶的不对称还原,特别优选为使用了来自乳酸杆菌属(lactobacillus sp.)的酮还原酶作为酶的不对称还原。使用酮还原酶的不对称还原利用酮还原酶、辅酶和辅酶再生体系来进行。酮还原酶的辅酶的典型例子中包含nadp。另外,作为使辅酶nadp再生的辅酶再生体系的典型例子,已知有利用葡萄糖脱氢酶(gdh)进行的葡萄糖的氧化。另外,使用酮还原酶的不对称还原优选在溶剂中、在缓冲液的存在下进行。
[0101]
例如,在使用化学催化剂等的不对称还原中,相对于1摩尔当量的式(a-6)所表示
的化合物,不对称还原中的还原剂的使用量通常为1.0~2.2摩尔当量,优选为1.2~2.0摩尔当量。
[0102]
在使用了酶的不对称还原中,相对于1g的式(a-6)的化合物,酶的使用量通常为1.0~25倍量,优选为5~20倍量,更优选为10倍量。
[0103]
在使用了来自乳酸杆菌属(lactobacillus sp.)的酮还原酶的不对称还原中,相对于1g的式(a-6)的化合物,酶的使用量通常为1.0~25倍量,优选为5~20倍量,更优选为10倍量。
[0104]
在使用了酶的不对称还原中可使用d-葡萄糖。在使用d-葡萄糖的情况下,相对于1g的式(a-6)的化合物,d-葡萄糖的使用量通常为1.0~5.0倍量,优选为1.5~3.5倍量,更优选为2.0倍量。
[0105]
在使用了酶的不对称还原中可使用葡萄糖脱氢酶(gdh)。在使用葡萄糖脱氢酶(gdh)的情况下,相对于1g的式(a-6)的化合物,葡萄糖脱氢酶(gdh)的使用量通常为0.01~0.5倍量,优选为0.05~0.2倍量,更优选为0.05倍量或0.2倍量。
[0106]
在使用了酶的不对称还原中可使用辅酶,例如可使用烟酰胺腺嘌呤二核苷酸磷酸(nadp)。在使用烟酰胺腺嘌呤二核苷酸磷酸(nadp)的情况下,相对于1g的式(a-6)的化合物,烟酰胺腺嘌呤二核苷酸磷酸(nadp)的使用量通常为0.01~0.5倍量,优选为0.025~0.1倍量,更优选为0.025倍量或0.1倍量。
[0107]
不对称还原可在溶剂的存在下进行。作为溶剂,例如可使用:甲醇、乙醇、丙醇、丁醇等醇系溶剂;庚烷、己烷、辛烷、甲苯等烃系溶剂;四氢呋喃、1,4-二噁烷、丁醚等醚系溶剂;乙腈、二甲基亚砜、二甲基甲酰胺等极性溶剂;水等不参与反应的溶剂或它们的混合溶剂,可根据所用的酶的种类适当选择。
[0108]
在使用了酶的不对称还原中,作为缓冲液,例如可使用:磷酸缓冲液、磷酸钾缓冲液(例如,可由k2hpo4・
3h2o、kh2po4等试剂进行调制)、tris/hcl缓冲液、四硼酸钠/盐酸缓冲液、或三乙醇胺缓冲液等缓冲液,可根据所用的酶的种类适当选择。
[0109]
在使用了来自乳酸杆菌属(lactobacillus sp.)的酮还原酶的不对称还原中,作为溶剂,优选为二甲基亚砜、甲苯、水或它们的混合溶剂,更优选为甲苯、水或甲苯-水的混合溶剂。
[0110]
在使用了酶的不对称还原中,相对于1g的式(a-6)的化合物,有机溶剂的使用量通常为1.0~15倍量,优选为2~13倍量,更优选为5.0倍量。
[0111]
在使用了酶的不对称还原中,相对于1g的式(a-6)的化合物,缓冲液的使用量通常为10~40倍量,优选为15~30倍量,更优选为30倍量。
[0112]
在使用了酶的不对称还原中,反应溶液的ph通常为6.0~7.5,优选为6.0~6.5、6.5~7.0或6.0~7.0,更优选为6.0~7.0。
[0113]
进行不对称还原时的反应温度例如可从-78℃~溶剂回流的温度的范围、-78℃~室温的范围、0℃~溶剂回流的温度的范围或0℃~室温的范围等反应温度中适当选择。优选为0℃~室温的范围。
[0114]
进行使用了酶的不对称还原时的反应温度通常为酶不失活的温度范围,优选为20~60℃的范围,更优选为20~25℃的范围或50~60℃的范围,进一步优选为20~25℃的范围。
[0115]
《由式(a-7)制造式(b)的化合物或其盐的工序》式(b)的化合物或其盐可如下得到:通过将式(a-7)的手性醇化合物的叔丁氧羰基进行脱保护、或对将叔丁氧羰基脱保护而得到的式(b)的化合物的盐进行脱盐、或对将叔丁氧羰基脱保护而得到的式(b)的化合物转化成其盐。
[0116]
作为用于叔丁氧羰基的脱保护的试剂,通常可举出酸性试剂,但优选为氯化氢(盐酸、或使用乙酰氯与甲醇、乙醇、丙醇等醇系溶剂在溶剂体系中产生等)、溴化氢、三氟乙酸,更优选为氯化氢(盐酸、或使用乙酰氯与甲醇、乙醇、丙醇等醇系溶剂在溶剂体系中产生等)、三氟乙酸,特别优选为氯化氢(盐酸、或使用乙酰氯与甲醇、乙醇、丙醇等醇系溶剂在溶剂体系中产生等)。
[0117]
叔丁氧羰基的脱保护可在溶剂的存在下进行。作为进行叔丁氧羰基的脱保护时的溶剂,例如可举出:二氯甲烷、氯仿、1,2-二氯乙烷等卤素系溶剂;甲醇、乙醇、丙醇、丁醇等醇系溶剂;庚烷、己烷、辛烷、甲苯等烃系溶剂;四氢呋喃、1,4-二噁烷、丁醚等醚系溶剂;丙酮、乙腈、二甲基亚砜、二甲基甲酰胺等极性溶剂;水等不参与反应的溶剂或它们的混合溶剂,优选为二氯甲烷、氯仿、1,2-二氯乙烷等卤素系溶剂;甲醇、乙醇、丙醇、丁醇等醇系溶剂,更优选为丙醇(正丙醇)。
[0118]
进行叔丁氧羰基的脱保护时的反应温度例如可从-78℃~溶剂回流的温度的范围、-78℃~室温的范围、0℃~溶剂回流的温度的范围或0℃~室温的范围等反应温度中适当选择。优选为0~55℃的范围。
[0119]
式(b)的化合物的盐可使用碱进行脱盐。作为使式(b)的化合物的盐进行脱盐时的碱,可使用:碳酸氢钠、碳酸钾、碳酸钠、三乙胺、n,n-二异丙基乙胺、吡啶等碱,优选为碳酸氢钠、碳酸钾和碳酸钠,更优选为碳酸氢钠。
[0120]
式(b)的化合物的盐的脱盐可在溶剂的存在下进行。作为使式(b)的化合物的盐进行脱盐时的溶剂,例如可举出:二氯甲烷、氯仿、1,2-二氯乙烷等卤素系溶剂;四氢呋喃、1,4-二噁烷、丁醚等醚系溶剂;乙酸乙酯、乙酸异丙酯、乙腈、二甲基亚砜、二甲基甲酰胺等极性溶剂;水等不参与反应的溶剂或它们的混合溶剂,优选为乙酸乙酯、乙酸异丙酯、水、乙酸乙酯-水或乙酸异丙酯-水的混合溶剂,更优选为乙酸乙酯-水的混合溶剂。
[0121]
《由式(b)的化合物制造其盐的工序》可使用有机酸或无机酸使式(b)的化合物转化成盐。作为使式(b)的化合物转化成盐时的酸,例如可使用:盐酸、氢溴酸、氢碘酸、硝酸、硫酸、磷酸、甲酸、乙酸、三氟乙酸、丙酸、丁酸、戊酸、庚酸、癸酸、肉豆蔻酸、棕榈酸、硬脂酸、乳酸、山梨酸、扁桃酸、草酸、丙二酸、琥珀酸、富马酸、马来酸、苹果酸、酒石酸、枸橼酸、苯甲酸、水杨酸、邻苯二甲酸、肉桂酸、羟基乙酸、丙酮酸、含氧酸(oxylic acid,羟基酸)、水杨酸、n-乙酰半胱氨酸、甲磺酸、苯磺酸、对甲苯磺酸、天冬氨酸、谷氨酸等酸,优选为盐酸、氢溴酸、硝酸、硫酸、磷酸、乙酸、邻苯二甲酸、富马酸、草酸、酒石酸、马来酸、枸橼酸、琥珀酸、甲磺酸、对甲苯磺酸,更优选为盐酸、氢溴酸,进一步优选为氢溴酸。
[0122]
在使式(b)的化合物转化成盐时可在溶剂的存在下进行。作为使式(b)的化合物转化成盐时的溶剂,例如可举出:二氯甲烷、氯仿、1,2-二氯乙烷等卤素系溶剂;四氢呋喃、1,4-二噁烷、丁醚等醚系溶剂;甲醇、乙醇等醇系溶剂;乙酸乙酯、乙酸异丙酯、乙腈、二甲基亚砜、二甲基甲酰胺等极性溶剂;水等不参与反应的溶剂或它们的混合溶剂,优选为二氯甲
烷、氯仿、1,2-二氯乙烷、四氢呋喃、1,4-二噁烷、甲醇、乙醇、乙酸乙酯、水等不参与反应的溶剂或它们的混合溶剂,更优选为乙酸乙酯、水或乙酸乙酯-水。
[0123]
《制造式(a8)所表示的化合物的工序》式(a8)所表示的化合物是通过使式(sm8)所表示的酮化合物进行不对称还原而得到。
[0124]
作为不对称还原,例如可举出:使用化学催化剂等的不对称还原、使用了生物催化剂(酵母、真菌、霉菌、酶等)的不对称还原等。优选为使用了酶的不对称还原,更优选为使用了酮还原酶(kred:keto reductase)作为酶的不对称还原,特别优选为使用了来自escherichia coli sp. (大肠杆菌属)的酮还原酶作为酶的不对称还原。使用酮还原酶的不对称还原利用酮还原酶、辅酶和辅酶再生体系来进行。酮还原酶的辅酶的典型例子中包含nadp。另外,作为使辅酶nadp再生的辅酶再生体系的典型例子,已知有利用葡萄糖脱氢酶(gdh)进行的葡萄糖的氧化。另外,使用酮还原酶的不对称还原优选在溶剂中、在缓冲液的存在下进行。
[0125]
例如,在使用化学催化剂等的不对称还原中,相对于1摩尔当量的式(sm8)所表示的化合物,不对称还原中的还原剂的使用量通常为1.0~2.2摩尔当量,优选为1.2~2.0摩尔当量。
[0126]
在使用了酶的不对称还原中,相对于1g的式(sm8)所表示的化合物,酶的使用量通常为0.01~0.1倍量,优选为0.02~0.07倍量,更优选为0.047~0.05倍量。
[0127]
在使用了来自escherichia coli sp.的酮还原酶(kred)的不对称还原中,相对于1g的式(sm8)所表示的化合物,酶的使用量为0.01~0.1倍量,优选为0.02~0.07倍量,更优选为0.047~0.05倍量。
[0128]
在使用了酶的不对称还原中可使用d-葡萄糖。在使用d-葡萄糖的情况下,相对于1g的式(sm8)所表示的化合物,d-葡萄糖的使用量通常为1.0~5.0倍量,优选为1.5~3.5倍量,更优选为1.9~2.0倍量。
[0129]
在使用了酶的不对称还原中可使用葡萄糖脱氢酶(gdh)。在使用葡萄糖脱氢酶(gdh)的情况下,相对于1g的式(sm8)所表示的化合物,葡萄糖脱氢酶(gdh)的使用量通常为0.01~0.1倍量,优选为0.01~0.05倍量,更优选为0.019~0.02倍量。
[0130]
在使用了酶的不对称还原中可使用烟酰胺腺嘌呤二核苷酸磷酸(nadp)。在使用烟酰胺腺嘌呤二核苷酸磷酸(nadp)的情况下,相对于1g的式(sm8)所表示的化合物,烟酰胺腺嘌呤二核苷酸磷酸(nadp)的使用量通常为0.001~0.1倍量,优选为0.005~0.05倍量,更优选为0.009~0.01倍量。
[0131]
不对称还原可在溶剂的存在下进行。作为溶剂,例如可使用:甲醇、乙醇、丙醇、丁醇等醇系溶剂;庚烷、己烷、辛烷、甲苯等烃系溶剂;四氢呋喃、1,4-二噁烷、丁醚等醚系溶剂;丙酮、乙腈、二甲基亚砜、二甲基甲酰胺等极性溶剂;水等不参与反应的溶剂或它们的混合溶剂,可根据所用的酶的种类适当选择。
[0132]
在使用了酶的不对称还原中,作为缓冲液,例如可使用:磷酸缓冲液、磷酸钾缓冲液(例如,可由k2hpo4・
3h2o、kh2po4等试剂进行调制)、tris/hcl缓冲液、四硼酸钠/盐酸缓冲液、或三乙醇胺缓冲液等缓冲液,可根据所用的酶的种类适当选择。
[0133]
在使用了来自escherichia coli sp.的酮还原酶(kred)的不对称还原中,作为溶
剂,优选为二甲基亚砜、水或二甲基亚砜-水的混合溶剂。
[0134]
在使用了来自escherichia coli sp.的酮还原酶(kred)的不对称还原中,相对于1g的式(sm8)所表示的化合物,有机溶剂的使用量通常为1.0~10倍量,优选为2~5倍量,更优选为2.5~3.0倍量。
[0135]
在使用了来自escherichia coli sp.的酮还原酶(kred)的不对称还原中,相对于1g的式(sm8)所表示的化合物,缓冲液的使用量通常为10~40倍量,优选为15~30倍量,更优选为28~30倍量。
[0136]
在使用了酶的不对称还原中,反应溶液的ph通常为6.0~7.5,优选为6.5~7.0。
[0137]
在进行不对称还原时的反应温度例如可从-78℃~溶剂回流的温度的范围、-78℃~室温的范围、0℃~溶剂回流的温度的范围或0℃~室温的范围等反应温度中适当选择。优选为0℃~室温的范围。
[0138]
进行使用了酶的不对称还原时的反应温度通常为酶不失活的温度范围,优选为20~60℃的范围,更优选为20~35℃的范围,进一步优选为20~30℃的范围。
[0139]
本说明书中,只要没有特别说明,则在记载为式(sm8)的情况下,是指包含其下位的式(例如,式(sm8-fl)、式(sm8-cl)、式(sm8-br)、式(sm8-id)等)。此外,同样地,在本说明书中,只要没有特别说明,则在记载为式(a8)的情况下,是指包含其下位的式(例如,式(a8-fl)、式(a8-cl)、式(a8-br)、式(a8-id)等)。
[0140]
另外,式(sm8-fl)是式(sm8)所表示的化合物中x=氟原子的化合物。式(sm8-cl)是式(sm8)所表示的化合物中x=氯原子的化合物。式(sm8-br)是式(sm8)所表示的化合物中x=溴原子的化合物。式(sm8-id)是式(sm8)所表示的化合物中x=碘原子的化合物。
[0141]
另外,式(a8-fl)是式(a8)所表示的化合物中x=氟原子的化合物。式(a8-cl)是式(a8)所表示的化合物中x=氯原子的化合物。式(a8-br)是式(a8)所表示的化合物中x=溴原子的化合物。式(a8-id)是式(a8)所表示的化合物中x=碘原子的化合物。
[0142]
《由式(a8)制造式(b)所表示的化合物的工序》通过在金属催化剂存在下使用氨(氨水(例如,有25%、28%、30%等))与式(a8)所表示的化合物进行氨基化反应,得到式(b)所表示的化合物。氨水的浓度(%)为w/w%或w/v%。
[0143]
作为使用氨作为氮源的式(a8)所表示的化合物的氨基化反应的催化剂,例如可举出:pd催化剂、cu催化剂等。作为pd催化剂,例如可举出:pd2(dba)
3 pdcl
2-josiphos络合物等,作为cu催化剂,例如可举出:cui、cu(oac)2、cu2o、cuo、cubr、cucl、cuso4、cufe2o4等,优选为cu催化剂,更优选为cu2o。
[0144]
作为氨基化反应的溶剂,例如可举出:二甲基亚砜、n,n-二甲基甲酰胺、n-甲基吡咯烷酮(nmp)、1,4-二噁烷、乙腈、甲苯、它们的混合溶剂等溶剂,优选为n-甲基吡咯烷酮(nmp)。
[0145]
在氨基化反应中可存在碱,作为碱,例如可举出:碳酸钾、磷酸钾、碳酸铯、n,n-二异丙基乙胺、三乙胺等碱。
[0146]
氨基化反应使用封管反应容器(例如,可为不锈钢制、玻璃制等)加热封管而进行。通常,在进行加热反应的情况下,不会被加热到所用的溶剂或试剂的沸点以上,在所用的溶剂或试剂的沸点以上的温度下进行反应的情况下,使用封管反应容器等在密闭体系中进行
加热。
[0147]
可用于进行氨基化反应时的溶剂的例子及其沸点为:二甲基亚砜(沸点189℃)、n,n-二甲基甲酰胺(沸点153℃)、n-甲基吡咯烷酮(nmp) (沸点202℃)、1,4-二噁烷(沸点101℃)、乙腈(沸点82℃)、甲苯(沸点110.6℃)。此外,氨水的沸点是对于25%氨水为37.7℃、对于32%氨水为24.7℃。
[0148]
进行氨基化反应时的反应温度例如可从100℃~250℃的范围、100℃~200℃的范围、100℃~150℃的范围等反应温度中适当选择。优选为100~120℃的范围。
[0149]
氨基化反应中,在使用cu2o的情况下,相对于1摩尔当量的式(a8)所表示的化合物,金属催化剂的使用量通常为0.1~1.0摩尔当量,优选为0.2~0.8摩尔当量,更优选为0.5~0.7摩尔当量。
[0150]
氨基化反应中,相对于1g的式(a8)所表示的化合物,有机溶剂的使用量通常为0.1~30倍量,优选为0.5~20倍量。
[0151]
氨基化反应中,相对于1g的式(a8)所表示的化合物,氨水的使用量通常为1.0~50倍量,优选为2.5~30倍量,更优选为2.5~3.5倍量。
[0152]
只要没有特别说明,则本说明书中所记载的数值范围还包含该值的
±
10%的值。例如,在记载为0.1~1.0摩尔当量的情况下,是指0.1
±
0.01~1.0
±
0.1摩尔当量,在记载为0.1~30倍量的情况下,是指0.1
±
0.01~30
±
3倍量。
[0153]
《制造式(i)的化合物的工序》式(i)的化合物是通过使用dmt-mm作为缩合剂将式(b)的化合物或其盐与式(ca-1)的化合物进行缩合反应而得到。
[0154]
缩合反应可在溶剂的存在下进行。作为溶剂,例如可使用:甲醇、乙醇、丙醇、异丙醇、丁醇等醇系溶剂;四氢呋喃、1,4-二噁烷、丁醚等醚系溶剂;水等不参与反应的溶剂或它们的混合溶剂,优选为醇系溶剂、水、或它们的混合溶剂,更优选为甲醇、乙醇、异丙醇、水、或它们的混合溶剂,进一步优选为甲醇、乙醇或异丙醇,特别优选为甲醇或异丙醇。
[0155]
缩合反应中,相对于1摩尔当量的式(b)的化合物或其盐,式(ca-1)的羧酸化合物的使用量通常为0.5~2.0摩尔当量,优选为0.5~1.5摩尔当量,更优选为0.7~1.25摩尔当量。如后所述,本发明人发现了:通过使用dmt-mm作为缩合剂,式(ca-1)的化合物的羧基与式(b)的化合物的氨基选择性地进行缩合反应;因此,缩合反应中,无需对式(b)的化合物的羟基进行保护。
[0156]
缩合反应中,作为式(b)的化合物的盐,优选为hcl盐或hbr盐。
[0157]
缩合反应中,相对于1摩尔当量的式(b)的化合物或其盐,作为缩合剂的dmt-mm的使用量通常为1.0~2.0摩尔当量,优选为1.1~1.8摩尔当量,更优选为1.2~1.5摩尔当量。
[0158]
缩合反应中,在使用式(b)的化合物的盐的情况下,可添加碱。作为碱,例如可举出:三乙胺、n,n-二异丙基乙胺、吡啶等有机碱;氢氧化锂(氢氧化锂
・
一水合物)、氢氧化钠、氢氧化钾、碳酸锂、碳酸钠、碳酸钾等无机碱。优选为三乙胺、n,n-二异丙基乙胺、吡啶、碳酸钠或碳酸钾,更优选为三乙胺。
[0159]
缩合反应中,相对于1摩尔当量的式(b)的化合物的盐,在使用式(b)的化合物的盐的情况下可添加的碱的量通常为1.0~2.5摩尔当量,优选为1.05~2.3摩尔当量,更优选为1.05~2.1摩尔当量。
[0160]
缩合反应中,相对于1g的式(b)的化合物或其盐,溶剂的使用量通常为5.0~100倍量,优选为5~40倍量,更优选为5~30倍量。
[0161]
进行缩合反应时的反应温度例如可从-78℃~溶剂回流的温度的范围、-78℃~室温的范围、0℃~溶剂回流的温度的范围或0℃~室温的范围等反应温度中适当选择。优选为0℃~室温的范围。
[0162]
上述方案[1]中的式(a)的8-氨基-1,2,3,4-四氢萘-2-醇[cas no.624729-66-4]可通过文献已知的制造方法、例如国际公开第2009/050289号小册子(专利文献6)中记载的下述制造方法,以8-氨基萘-2-醇(式(sm-1))作为起始物质,将萘环进行选择性地还原而制造。
[0163]
[化学式81](图解4-1)上述方案[7]~[11]和上述方案[17]~[20]中的式(ca-1)所表示的(e)-2-(7-(三氟甲基)色满-4-亚基)乙酸[cas no.920334-15-2]可通过文献已知的制造方法、例如国际公开第2007/010383号小册子(专利文献1)中记载的下述制造方法,以3-羟基三氟甲苯(3-hydroxybenzotrifluoride) (式(ca-sm))作为起始物质,经过多个工序而制造。
[0164]
[化学式82](图解4-2)上述方案[13]、[15]、[17]和[18]中的式(sm8)所表示的化合物可使用市售化合物。或者,可使用市售化合物通过文献已知的制造方法而获取。
[0165]
式(sm8)所表示的化合物中x=氟原子的化合物(式(sm8-fl))可通过例如欧州专利公开第343830号小册子中记载的下述(图解4-3)的制造方法进行制造。
[0166]
[化学式83](图解4-3)
式(sm8)所表示的化合物中x=氯原子的化合物(式(sm8-cl))可通过例如欧州专利公开第343830号小册子中记载的下述(图解4-4)的制造方法进行制造。
[0167]
[化学式84](图解4-4)式(sm8)所表示的化合物中x=溴原子的化合物(式(sm8-br))可通过例如journal of medicinal chemistry、36(17)、第2485-93页、1993年中记载的下述(图解4-5)的制造方法进行制造。
[0168]
[化学式85](图解4-5)尚需说明的是,式(sm8)中x=碘原子的化合物(式(sm8-id))可按照上述式(sm8-fl)、式(sm8-cl)和式(sm8-br)的制造方法进行制造(图解4-6)。
[0169]
[化学式86](图解4-6)
制造方法中的各工序的原料化合物还可直接以反应液的形式或以粗产物的形式用于下一个工序。此外,也可按照常规方法从反应混合物中分离,其本身可通过已知的手段、例如萃取、浓缩、中和、过滤、蒸馏、重结晶、色谱等分离手段容易地进行纯化。
[0170]
在上述反应中使用混合溶剂的情况下,还可将两种以上的溶剂以适当的比例、例如按照容量比或重量比计为1:1~1:10的比例进行混合而使用。
[0171]
对制造方法的各工序的反应时间没有特别限定,只要是反应充分进行的时间则没有限定。例如,反应时间可以是0.1小时、0.5小时、1小时、1.5小时、2小时、3小时、4小时、5小时、10小时、12小时、18小时、24小时、36小时、48小时、60小时、72小时或115小时的各时间和以它们作为下限值和上限值的范围的时间。
[0172]
上述反应温度中,例如
“‑
78℃~溶剂回流的温度的范围”的意思是指从-78℃到反应中所用的溶剂(或混合溶剂)回流的温度为止的范围内的温度。例如,在使用甲醇作为溶剂的情况下,
“‑
78℃~溶剂回流的温度”是指从-78℃到甲醇回流的温度为止的范围内的温度。
[0173]“0℃~溶剂回流的温度”也同样,是指从0℃到反应中所用的溶剂(或混合溶剂)回流的温度为止的范围内的温度。该温度的下限值如上所述例如为-78℃或0℃,但也可以是其它20℃、23℃、25℃、40℃、50℃、70℃、80℃、90℃、100℃、或150℃等的温度。
[0174]
上述反应温度中,反应温度的下限值和上限值可以是例如各温度的
±
1℃、
±
2℃、
±
3℃、
±
4℃、
±
5℃的温度。
[0175]
本说明书的制造方法中,只要没有特别说明,则“室温”是指实验室、研究室等的温度,本说明书的实施例中的“室温”是显示通常约1℃~约30℃的温度(日本药典规定)。显示优选为通常约5℃~约30℃的温度,更优选为通常约15℃~约25℃的温度,进一步优选为20
±
3℃的温度。
[0176]
本说明书中的化合物根据取代基的种类有时会形成酸加成盐。作为所涉及的盐,只要是制药学上可接受的盐,则没有特别限定,例如可举出:与无机酸的盐、或与有机酸的盐等。作为与无机酸的盐的适当例子,例如可举出:与盐酸、氢溴酸、氢碘酸、硝酸、硫酸、磷酸等的盐。作为与有机酸的盐的适当例子,例如可举出:与甲酸、乙酸、三氟乙酸、丙酸、丁酸、戊酸、庚酸、癸酸、肉豆蔻酸、棕榈酸、硬脂酸、乳酸、山梨酸、扁桃酸等脂族一元羧酸等的盐;与草酸、丙二酸、琥珀酸、富马酸、马来酸、苹果酸、酒石酸等脂族二元羧酸的盐;与枸橼酸等脂族三元羧酸的盐;与苯甲酸、水杨酸等芳族一元羧酸的盐;与邻苯二甲酸等芳族二元羧酸的盐;与肉桂酸、羟基乙酸、丙酮酸、含氧酸、水杨酸、n-乙酰半胱氨酸等有机羧酸的盐;与甲磺酸、苯磺酸、对甲苯磺酸等有机磺酸的盐;与天冬氨酸、谷氨酸等酸性氨基酸类的酸
加成盐。
[0177]
上述盐可按照常规方法、例如在通过混合本说明书中的化合物和包含适量酸的溶液而形成目标盐之后,通过分步进行过滤收集、或者馏去该混合溶剂而得到。此外,本说明书中的化合物或其盐可与水、乙醇、甘油等溶剂形成溶剂合物。作为有关盐的综述,出版了handbook of pharmaceutical salts:properties, selection, and use. stahl & wermuth (wiley-vch, 2002年),在本书中进行了详细的记载。
[0178]
如下述(图解5)所示,式(b)的化合物或其盐能够以式(a)的化合物作为起始物质,经由式(a-5)、式(a-6)和式(a-7)的化合物进行制造。
[0179]
[化学式87](图解5)此外,如下述(图解6)所示,式(b)的化合物或其盐可通过将式(a)的化合物的氨基的保护基变更为叔丁氧羰基以外的保护基、例如甲氧羰基、乙氧羰基、苄氧羰基、2,2,2-三氯乙氧羰基、9-芴基甲氧基羰基或烯丙氧基羰基等氨基甲酸酯系的保护基;甲磺酰基、乙磺酰基、苯磺酰基、甲苯基或硝基苯磺酰基等磺酰基系的保护基;乙酰基、乙基羰基、三氟乙酰基或苯甲酰基等烷基羰基系或芳基羰基系保护基等的保护基p1,按照上述(图解5)的方法来制造。
[0180]
[化学式88](图解6)
尚需说明的是,关于式(a)的化合物的保护基p1的保护或式(a-7p)的化合物的保护基p1的脱保护,例如可根据保护基p1的种类,从文献已知的方法、例如“有机合成中的保护基(protective groups in organic synthesis, 4th edition) 第4版、2007年、john wiley&sons、 greene等人”的出版物(成書)中记载的保护/脱保护法中选择条件。
[0181]
如下述(图解7)所示,式(b)所表示的化合物能够以式(sm8)所表示的化合物作为起始物质,经由式(a8)所表示的化合物进行制造。
[0182]
[化学式89](图解7)此外,如下述(图解8)所示,式(b)所表示的化合物能够以式(sm8-br)所表示的化合物作为起始物质,经由式(a8-br)所表示的化合物进行制造。
[0183]
[化学式90](图解8)
如下述(图解9)中所示,式(i)所表示的化合物能够以式(a)所表示的化合物作为起始物质,经由式(b)所表示的化合物进行制造。(图解9)中,式(b-ha)所表示的化合物表示式(b)所表示的化合物的酸ha的盐,ha表示酸。
[0184]
[化学式91](图解9)另外,如下述(图解10)中所示,式(i)所表示的化合物能够以式(a)所表示的化合物作为起始物质,经由式(b)所表示的化合物进行制造。(图解10)中的取代基p1的定义与上述(图解6)中的定义相同。(图解10)中,式(b-ha)所表示的化合物表示式(b)所表示的化合物的酸ha的盐,ha表示酸。
[0185]
[化学式92](图解10)
另外,如下述(图解11)中所示,式(i)所表示的化合物能够以式(sm8)所表示的化合物作为起始物质,经由式(b)所表示的化合物进行制造。(图解11)中,式(b-ha)所表示的化合物表示式(b)所表示的化合物的酸ha的盐,ha表示酸。
[0186]
[化学式93](图解11)另外,如下述(图解12)中所示,式(i)所表示的化合物能够以式(sm8-br)所表示的化合物作为起始物质,经由式(b)所表示的化合物进行制造。(图解12)中,式(b-ha)所表示的化合物表示式(b)所表示的化合物的酸ha的盐,ha表示酸。
[0187]
[化学式94](图解12)
本说明书中,在作为外消旋体的式(a)、式(a-5)、式(a-5p)的化合物中包含(r)型和(s)型。例如,式(a-5)中是指包含式(a-5s)和式(a-5r) (=式(a-7))。
[0188]
[化学式95][酮的不对称还原]作为将位于分子内的酮基转化成手性醇基的方法,已知有各种反应。例如有下述方法:使用还原剂(硼氢化钠、氢化锂铝(lah)、硼烷-四氢呋喃(bh3·
thf)等)将酮基转化成外消旋的醇基,之后通过分步重结晶法(使光学拆分剂与外消旋体进行离子键合而得到结晶性的非对映异构体。将其通过重结晶进行分离,根据需要进行中和,由此得到游离的手性化合物的方法)、非对映异构体法(参照国际公开第2009/055749号小册子)、手性柱法(参照国际公开第2009/050289号小册子)等方法衍生成手性醇基。
[0189]
此外,还已知使用过渡金属催化剂(例如ru、rh等)的不对称还原反应(国际公开第2009/050289号小册子、organometallics 10、第500页~、1991年等)、将al(ch3)3与作为配体的binol组合的不对称还原反应(angew. chem. int. ed.、41、第1020页~、2002年)、使用了手性的ru (binap)催化剂的不对称还原反应(j. am. chem. soc. 110、第629页~、1988年)、使用噁唑硼烷(oxazaborolidine)的不对称还原反应(j. am. chem. soc. 109、第5551页~、1987年)、使用了生物催化剂(酵母、真菌、霉菌、酶等)的不对称还原反应(参照表1)等。
[0190]
在几个方案中,不对称还原优选基于生物催化剂的不对称还原。基于生物催化剂的不对称还原不仅具有立体选择性高、可使用有机溶剂和/或水作为反应溶剂、在温和的条件(常温、常压)下进行反应、较化学催化剂廉价等优点,还能够减少反应后的废弃物,能够成为环境友好型的反应,因此近年来成为备受关注的反应,是用于容易地得到手性化合物的有效反应。
[0191]
通常,在使用酶的不对称还原反应中,所得的手性化合物的化学收率(%)和光学活性收率(ee%)会依赖于反应特异性(酶特有的对反应种类的选择性)、底物特异性(对底物种类的选择性)、反应条件(反应温度、ph值、溶剂、反应时间等)而发生变化。对于许多酶而言,反应特异性非常高,且一种酶所催化的反应是有限的,但有各种底物特异性较高的酶~不高的酶。因此,例如在将酮基不对称还原成手性醇基的情况下,即使选择在与所用的底物(酮化合物)具有类似结构的化合物中可得到良好的化学收率和光学活性收率的酶在同样的条件下进行酶反应,也未必能够以同样的化学收率和光学活性收率得到目标的手性醇化合物。
[0192]
例如,作为可将β-四氢萘酮[cas编号:530-93-8]的酮基选择性地还原成手性醇的生物催化剂,已知有表1的各种物质。
[0193]
[表1-1]
[表1-2]
[流动化学]通常,合成反应中有流动法(流动化学)和分批法。流动化学是指使用了下述反应装置的连续合成法,所述反应装置从装有2种以上的异种溶液(例如,原料+溶剂、试剂+溶剂等)的容器开始,使用泵以某种恒定的流速,使所述溶液通过管,输送到反应容器、然后输送到回收桶(recovery drum)中。
[0194]
可将流动化学用于通过氧化反应将式(a-5)的化合物转化成式(a-6)的化合物时。该流动化学中使用的反应装置的例子示于图1中。图1所示的反应装置包括:氮导入口(l1、l2、l3、l4);包含原料、tempo和二氯甲烷的容器(m1);包含kbr、nahco3和水的容器(m2);包含5.0wt%naclo的容器(m3);泵(p1、p2、p3);预冷管(t1、t2、t3);搅拌机(s1、s2、s3);以及反应容器(r1、r2、r3)。
[0195]
图1的反应装置例如以下述方式进行使用。首先,在容器m1中装入原料(式(a-5)的化合物)、tempo和二氯甲烷,在容器m2中装入kbr、碳酸氢钠和水,在容器m3中装入5.0wt%naclo。从各氮导入口l1、l2、l3、l4流入氮气,同时使用各泵p1、p2和p3,使各试剂以各自规定的流速从各容器m1、m2和m3流入,通过预冷管t1、t2和t3,再依次通过反应容器r1、反应容器r2和反应容器r3,注入回收桶cd中。然后,从回收桶cd得到目标物(式(a-6)的化合物)。
[0196]
流动化学还可适用于在通常的分批法中难以确保安全性的这样的反应(关于流动
化学的综述参照chemsuschem, 5(2), special issue;flow chemistry, 第213-439页, 2012年2月13日)。
[0197]
分批法是通常的合成反应,是指在使用反应容器进行反应后纯化产物的方法。分批法具有可经过多个工序合成化合物的优点。
[0198]
流动法(流动化学)例如使用连续搅拌槽反应器(cstr:continuous stirred tank reactor)作为其反应装置,采用流动模式进行反应。流动法可使用小的反应容器进行反应,故反应效率高,此外,还可精密地控制反应条件,可实现目标物的稳定供给。
[0199]
[氨基化反应]将卤化芳基的卤素原子转换成氨基的方法(氨基化反应),可在金属催化剂存在下、在配体(ligand)的存在或不存在下,使用作为氮源的由nhrar
b (ra和rb各自独立表示氢原子、甲基、乙基、苄基等取代基)、rcconh
2 (rc独立表示甲基、乙基、苄基、甲氧基、乙氧基、叔丁氧基、苄氧基等取代基)等表示的化合物来进行。
[0200]
关于使用氨作为氮源的卤化芳基的氨基化反应,作为文献已知的方法,例如已知有使用以下的金属催化剂的方法,但不限于这些。pd2(dba)
3 (j. am. chem. soc., 129(34), 第10354~10355页, 2007年)、pdcl
2-josiphos络合物(j. am. chem. soc., 128(31), 第10028~10029页, 2006年)、cui (chem. commun., 26, 第3052~3054页, 2008年; j. org. chem., 74(12), 第4542
–
4546页, 2009年)、cu(oac)
2 (angew. chem. int. ed., 48(2), 第337-339页, 2009年)、cu2o (ukrainskii khimiche skii zhurnal (russian edition), 53(12), 第1299-302页, 1987年)。
[0201]
例如,在分子内存在仲醇的8-卤代-1,2,3,4-四氢萘-2-醇的氨基化反应,已知有使用了pd2(dba)3作为金属催化剂和使用了氨基甲酸叔丁酯作为氮源的氨基化反应,但使用其它金属催化剂的例子是未知的。
[0202]
在几个方案中,氨基化反应优选可利用氨直接导入氨基的反应。另外,例如在使用nhr
a1rb1 (r
a1
为氢原子,r
b1
表示苄基、4-甲氧基苄基等氨基的保护基)、rcconh
2 (rc独立表示甲基、乙基、苄基、甲氧基、乙氧基、叔丁氧基、苄氧基等取代基)等的保护氨基化合物进行取代之后,通过进行保护基的脱保护也可导入氨基。但是,由于使用保护氨基化合物的氨基化反应需要进行保护基的脱保护工序,因此在考虑大量合成或工业生产的情况下,优选可利用氨直接导入氨基的反应。
[0203]
[缩合反应]通常,关于具有羧基的化合物与具有氨基的化合物的缩合反应,例如,可通过使用1,3-二环己基碳二亚胺(dcc)、1-乙基-3-(3
’‑
二甲基氨基丙基)碳二亚胺盐酸盐(wsc
・
hcl)、苯并三唑-1-基氧基三(二甲基氨基)磷鎓六氟磷酸盐(bop试剂)、双(2-氧代-3-噁唑烷基)次膦酰氯(bop-cl)、2-氯-1,3-二甲基咪唑鎓六氟磷酸盐(cip)等缩合剂进行缩合反应,来形成酰胺键(例如,参照实验化学讲座第4版22有机合成iv酸
・
氨基酸
・
肽、第191-309页、1992年、丸善等)。
[0204]
作为研究缩合剂的结果,本发明人发现了:在同时具有羟基和苯胺性氨基的式(b)的化合物与具有羧基的式(ca-1)所表示的杂环亚基乙酸的缩合反应中,特别是通过使用dmt-mm作为缩合剂,可收率良好且容易地制造式(i)的化合物。
[0205]
本说明书中引用的所有出版物、例如现有技术文献和公开公报、专利公报以及其
它专利文献,其整体作为参照而纳入本说明书中。本说明书包含成为本技术的优先权要求的基础的中国专利申请号201910783254.8号(2019年8月23日申请)、国际专利申请号pct/jp2019/036451号(2019年9月18日申请)、中国专利申请号202010355546.4号(2020年4月29日申请)的权利要求书、说明书和附图的公开内容。
实施例
[0206]
以下,列举实施例来具体地说明本发明,但本发明并不受该实施例的任何限定。
[0207]
在式(a-5)、式(a-6)、式(a-7)和式(b)的化合物的核磁共振光谱(nmr)的测定中,使用了配备的bruker avanceiii 400 mhz nmr波谱仪(bruker 5mm pabbo z-梯度探针、topspin 3.5软件)。此外,在式(b)的化合物的溴酸盐和式(i)的化合物的核磁共振光谱(nmr)的测定中,使用了jeol jnm-la300 ft-nmr (日本电子)。
[0208]
式(a-5)、式(a-6)、式(a-7)和式(b)的化合物按照以下条件的高效液相色谱(hplc)进行测定。
[0209]
[表2]
[表3]
[表4][表5][表6]
[表7]此外,式(b)的化合物的溴酸盐和式(i)的化合物的液相色谱-质谱(lc-mass)通过以下的方法进行测定。
[0210]
[uplc]使用waters aquity uplc系统和beh c18柱(2.1mm
×
50mm、1.7μm) (waters),采用了乙腈:0.05%三氟乙酸水溶液=5:95 (0分钟)~95:5 (1.0分钟)~95:5 (1.6分钟)~5:95 (2.0分钟)的流动相和梯度条件。
[0211]1h-nmr数据中,在nmr信号的图案中,s为单峰,d为双峰,t为三重峰,q为四重峰,m为多重峰,brs为宽峰,j为偶联常数,hz为赫兹,dmso-d6为氘代二甲基亚砜,cdcl3为氘代氯仿。在1h-nmr数据中,对于羟基(oh)、氨基(nh2)、酰胺基(conh)的质子等因为是宽带而无法确认的信号没有记载在数据中。
[0212]
lc-mass数据中,m为分子量、rt为保留时间、[m+h]
+
为分子离子峰。
[0213]
(实施例1a~1d) 叔丁基(7-羟基-5,6,7,8-四氢萘-1-基)氨基甲酸酯(a-5)的合
成[化学式96](实施例1a)在式a的化合物(依据国际公开第2009/050289号小册子记载的制造方法制造的) (0.5g)和碳酸氢钠(0.154g)的1,4-二噁烷(5ml)-水(5ml)的溶液中加入二碳酸二叔丁酯(boc2o) (0.74g),在反应温度20~30℃下搅拌22小时。再加入碳酸氢钠(0.104g),在反应温度20~30℃下搅拌18小时。再加入二碳酸二叔丁酯(0.15g),在反应温度20~30℃下搅拌5小时。再加入二碳酸二叔丁酯(0.15g),在反应温度20~30℃下搅拌16小时。再加入二碳酸二叔丁酯(0.1g),在反应温度20~30℃下再搅拌1小时。在反应液中加入乙酸乙酯,分离有机层。将水层用乙酸乙酯进行洗涤,与先前得到的有机层合并,之后用饱和食盐水进行洗涤。将有机层减压浓缩,使所得残余物用二氯甲烷和正庚烷进行固化,得到了标记化合物(0.46g)。
[0214]
(实施例1b)在式a的化合物(依据国际公开第2009/050289号小册子记载的制造方法制造的) (1.0g)和碳酸氢钠(1.55g)的四氢呋喃(10ml)-水(10ml)的溶液中加入二碳酸二叔丁酯(boc2o) (1.61g),在反应温度45~55℃下搅拌17小时。再加入二碳酸二叔丁酯(0.13g),在反应温度45~55℃下搅拌2小时。将反应液冷却至室温,之后加入甲基叔丁基醚(mtbe),用10w/v%枸橼酸溶液调节至ph=5~6,分离有机层。将水层用甲基叔丁基醚进行萃取,与先前得到的有机层合并,之后用水和饱和食盐水进行洗涤。将有机层减压浓缩,使所得残余物用二氯甲烷和正庚烷进行固化,得到了标记化合物(1.34g)。
[0215]
(实施例1c)在式a的化合物(依据国际公开第2009/050289号小册子记载的制造方法制造的) (10g)和碳酸氢钠(15.5g)的四氢呋喃(100ml)-水(100ml)的溶液中加入二碳酸二叔丁酯(boc2o) (17.4g),在反应温度45~55℃下搅拌17小时。再加入二碳酸二叔丁酯(1.3g),在反应温度45~55℃下搅拌2小时。再加入二碳酸二叔丁酯(1.3g),在反应温度45~55℃下再搅拌1小时。将反应液冷却至室温,之后加入甲基叔丁基醚(mtbe),用10%枸橼酸溶液调节至ph=5~6,分离有机层。将水层用甲基叔丁基醚进行萃取,与先前得到的有机层合并,之后用水和饱和食盐水进行洗涤。将有机层减压浓缩,使所得残余物用二氯甲烷和正庚烷进行固化,得到了标记化合物(15.1g)。
[0216]
(实施例1d)将式a的化合物(依据国际公开第2009/050289号小册子记载的制造方法制造的) (218.5g)的四氢呋喃(1.9l)溶液调节至20~30℃的温度,在温度20~30℃下用10分钟加入碳酸氢钠水溶液(319g (3.2eq)的碳酸钠、1.9l的水)。将上述混合溶液的温度设为0~10
℃,边保持同一温度边用15分钟加入二碳酸二叔丁酯(413g)。将反应温度设为45~55℃,在同一温度下搅拌18小时。将反应温度冷却至20~30℃,之后在反应溶液中加入甲基叔丁基醚(1.9l),在20~30℃下搅拌10分钟。加入10%枸橼酸溶液(2.5l),分离有机层。将水层用甲基叔丁基醚(1l
×
2次)进行萃取,与先前得到的有机层合并,之后用水(1l
×
2次)进行洗涤。进行减压浓缩直至有机层达到约500ml,之后加入二氯甲烷(1l),进行减压浓缩直至有机层达到约500ml,上述操作进行2次,之后加入正庚烷(1l),进行减压浓缩直至有机层达到约500ml,加入正庚烷(800ml),进行减压浓缩直至有机层达到约600ml,加入二氯甲烷(300ml)和正庚烷(600ml),进行减压浓缩直至有机层达到约600ml,加入二氯甲烷(1l),加入活性炭(21g),将混合溶液在20~30℃下搅拌2小时。之后,过滤混合溶液,将滤液减压浓缩直至达到约500ml,加入二氯甲烷(800ml),进行减压浓缩直至溶液达到约500ml。加入二氯甲烷(800ml)进行过滤,由此得到了固体。将所得固体在35℃下干燥15小时,得到了作为灰黑色固体的标记化合物(303.3g)。
[0217]
[式(a-5)的化合物的物性数据](1h nmr、400 mhz、生产商:bruker、dmso-d6、δ ppm)(实施例2a-2f) 叔丁基(7-氧代-5,6,7,8-四氢萘-1-基)氨基甲酸酯(a-6)的合成[化学式97](实施例2a)进行与(实施例1a~1d)的方法同样的操作,使用由(r)-8-氨基-1,2,3,4-四氢萘-2-醇得到的叔丁基(r)-(7-羟基-5,6,7,8-四氢萘-1-基)氨基甲酸酯(式(a-7)) (0.5g),在溶剂(二氯甲烷(12.5ml)-水(7.5ml))、反应温度(0~5℃)下,在下表所示的试剂条件下进行氧化反应,确认得到了标记化合物(采用了hplc的ipc纯度(ipc=工序分析))。
[0218]
[表8]
(实施例2b)进行与(实施例1a~1d)的方法同样的操作,使用由(r)-8-氨基-1,2,3,4-四氢萘-2-醇得到的叔丁基(r)-(7-羟基-5,6,7,8-四氢萘-1-基)氨基甲酸酯(式(a-7)) (0.5g),在反应温度(0~5℃)下,在下表所示的试剂条件下进行氧化反应,确认得到了标记化合物(采用了hplc的ipc纯度)。
[0219]
[表9](实施例2c)进行与(实施例1a~1d)的方法同样的操作,将所得的叔丁基(r)-(7-羟基-5,6,7,8-四氢萘-1-基)氨基甲酸酯(式(a-7)) (10g)的二氯甲烷(250ml、25容量倍量)和水(150ml、15容量倍量)的溶液冷却至-2~2℃,在同一温度下加入tempo (0.5eq)、kbr (0.2eq)、nahco
3 (4.0eq)和naclo ((8.1%)、1.4eq.)。立即确认到在确认了ipc纯度处为95.5%纯度下反应结束。通过对反应溶液进行后处理,得到了标记化合物(1h-nmr收率为77.1%)。
[0220]
(实施例2d)实施了采用图1所示的连续搅拌槽反应器(cstr:continuousstirred tank reactor)的流动模式的tempo氧化。在容器m1中装入进行与(实施例1a~1d)的方法同样的
操作而得到的叔丁基-(7-羟基-5,6,7,8-四氢萘-1-基)氨基甲酸酯(式(a-5)) (25g)和tempo (0.5eq)的二氯甲烷溶液(500ml),在容器m2中装入kbr (0.05eq)、碳酸氢钠(4eq)和水(375ml),在容器m3中装入5.0wt%的naclo (1.3eq)。从各氮导入口l1、l2、l3、l4流入氮气,同时使用各泵p1、p2和p3,从各容器m1、m2和m3分别以13.67ml/分钟、9.83ml/分钟、4.27ml/分钟的流速流入各试剂,使其通过预冷管t1、t2和t3 (温度0~5℃、管长:2m、管径:1/16ss),再依次通过反应容器r1、反应容器r2、反应容器r3 (反应容器1、2和3为25ml容量,各反应容器冷却至0~5℃),注入到回收桶cd中(在各反应容器中的反应时间为0.9分钟)。以回收桶cd中得到的反应溶液的ipc纯度=96.5%得到了目标物。
[0221]
(实施例2e)实施了使用图1所示的连续搅拌槽反应器(cstr:continuous stirredtank reactor)的流动模式的tempo氧化。在容器m1中装入进行与(实施例1a~1d)的方法同样的操作而得到的叔丁基(r)-(7-羟基-5,6,7,8-四氢萘-1-基)氨基甲酸酯(式(a-5)) (35g)和tempo (0.5eq)的二氯甲烷(700ml),在容器m2中装入kbr (0.05eq)、碳酸氢钠(4eq)和水(525ml),在容器m3中装入5.0wt%的naclo (1.3eq)。从各氮导入口l1、l2、l3、l4流入氮气,同时使用各泵p1、p2和p3,从各容器m1、m2和m3分别以13.67ml/分钟、9.83ml/分钟、4.27ml/分钟的流速流入各试剂,使其通过预冷管t1、t2和t3 (温度0~5℃、管长:2m、管径:1/16ss),再依次通过反应容器r1、反应容器r2、反应容器r3 (反应容器1、2和3为25ml容量、各反应容器冷却至0~5℃),注入到回收桶cd中(在各反应容器中的反应时间为0.9分钟,流动结束需要47分钟)。回收桶cd中得到的反应溶液(850g)的ipc纯度为95.0%。在反应溶液中加入na2s2o4水溶液(na2s2o4:10g、水:250ml),搅拌30分钟。将有机层与水层分离,之后将有机层用水(300ml
×
2次)进行洗涤。减压下将有机层浓缩至1.5~2.5v的容量,之后加入正庚烷(30~50ml),在室温下搅拌1小时,再加入正庚烷(200ml),在室温下搅拌16小时。过滤收集所生成的固体,用正庚烷(70ml)进行洗涤,由此得到了作为灰白色固体的标记化合物(25.2g)。
[0222]
(实施例2f)实施了使用图1所示的连续搅拌槽反应器(cstr)的流动模式的tempo氧化。在容器m1中装入进行与(实施例1a~1d)的方法同样的操作而得到的叔丁基-(7-羟基-5,6,7,8-四氢萘-1-基)氨基甲酸酯(式(a-5)) (276.6g)和tempo (82.588g、0.5eq)的二氯甲烷(5535ml、20v),在容器m2中装入kbr (6.251g)、碳酸氢钠(352.943g)和水(4149ml),在容器m3中装入5.0wt%的naclo (1.849l)。从各氮导入口l1、l2、l3、l4流入氮气,同时使用泵p1、p2、p3,从m1、m2和m3分别以13.67ml/分钟、9.83ml/分钟、4.27ml/分钟的流速流入各试剂,使其通过预冷管t1、t2和t3 (温度0~5℃、管长:2m、管径:1/16ss),再依次通过反应容器r1、反应容器r2、反应容器r3 (反应容器1、2和3为25ml容量、各反应容器冷却至0~5℃),注入到回收桶cd中(流动模式下在各容器中的反应时间为0.9分钟,流动结束需要410分钟)。加入6845g所得的反应混合液、3.7%的na2s2o4溶液(2597g),搅拌30分钟。将有机层与水层进行分离,之后将有机层用水(3l
×
2次)进行洗涤。减压下将有机层浓缩至2.5v的容量,之后加入正庚烷(205ml)和已获取的式a-6的化合物的1个碎片,在室温下搅拌1小时。再加入正庚烷(2.1l),过滤收集所生成的固体,用正庚烷(800ml)进行洗涤,减压下干燥13小时,由此得到了作为茶色固体的标记化合物(190g)。
[0223]
[式(a-6)的化合物的物性数据](1h nmr、400 mhz、生产商:bruker、cdcl3、δ ppm)(实施例3a-3g) 叔丁基(r)-(7-羟基-5,6,7,8-四氢萘-1-基)氨基甲酸酯(a-7)的合成[化学式98](实施例3a)在玻璃烧瓶(容量为8ml)中混合kred (来自lactobalius sp.的酮还原酶、2.0g)、d-葡萄糖(0.2g)、葡萄糖脱氢酶(gdh) (0.02g)、烟酰胺腺嘌呤二核苷酸磷酸(nadp) (0.01g)和磷酸缓冲液(将21.25g的k2hpo4、10.62g的kh2po4加入到1000ml水中而调制的缓冲液:3.0ml)进行搅拌,调制了混合溶液a。将进行与(实施例2a-2f)的方法同样的操作而得到的式(a-6)的化合物(0.1g)溶解于二甲基亚砜(dmso) (0.2ml)中而得的混合溶液加入到先前调制的混合溶液a中。在反应温度23℃ (20~25℃)下搅拌43小时(在250rpm下、在回旋振荡器中振荡)。取出一部分反应溶液,进行hplc分析,由此确认得到了标记化合物。
[0224]
尚需说明的是,(实施例3a-3g)中使用的kred是来自lactobalius sp.的酮还原酶(enzymeworks, inc.、产品编号:ew-kred-172)。
[0225]
(实施例3b)在反应容器中混合kred (来自lactobalius sp.的酮还原酶、20g)、d-葡萄糖(2g)、葡萄糖脱氢酶(gdh) (0.2g)、烟酰胺腺嘌呤二核苷酸磷酸(nadp) (0.1g)和缓冲液(将0.86g的k2hpo4·
3h2o、0.3g的kh2po4加入到30ml水中而得的缓冲液)进行搅拌,调制了混合溶液b。将进行与(实施例2a-2f)的方法同样的操作而得到的式(a-6)的化合物(1g)溶解于甲苯(13ml)中而得的混合溶液加入到先前调制的混合溶液b中。在反应温度23℃ (20~25℃)下搅拌15小时。将反应溶液进行硅藻土过滤,分离水层和有机层,将水层用甲苯(30ml)进行萃取,与先前得到的有机层合并,之后用水(30ml
×
2次)进行洗涤,之后将有机层进行浓缩,由此得到了作为褐色油的标记化合物(0.5g、光学纯度为99.9%ee)。
[0226]
(实施例3c)在下表所示的反应条件下,实施了kred量和ph的研究。式(a-6)的化合物是进行与(实施例2a-2f)的方法同样的操作而得到的化合物。与实施例3b同样地,缓冲液由k2hpo4・
3h2o、kh2po4进行调制。
[0227]
[表10](实施例3d)在下表中记载的反应条件下,实施了溶剂量的研究。式(a-6)的化合物是进行与(实施例2a-2f)的方法同样的操作而得到的化合物。与实施例3b同样地,缓冲液由k2hpo4・
3h2o、kh2po4进行调制。
[0228]
[表11](实施例3e)在下表中所记载的反应条件下,实施了缓冲液量、原料量和krde量的研究。式(a-6)的化合物是进行与(实施例2a-2f)的方法同样的操作而得到的化合物。与实施例3b同样地,缓冲液由k2hpo4・
3h2o、kh2po4进行调制。
[0229]
[表12](实施例3f)使用进行与(实施例2a-2f)的方法同样的操作而得到的式(a-6)的化合物(10g)、甲苯(50ml)、缓冲液(300ml、k2hpo4・
3h2o和kh2po4的组成与上述实施例相同)、kred (100g)、d-葡萄糖(20g)、nadp (0.25g)和gdh (0.5g),在反应溶液的ph=6.0~7.0、反应温度23℃ (20~25℃)下连续进行23小时的酶反应,之后在50~60℃下进行16小时的酶反应,之后依据上述的后处理方法进行后处理,由此得到了作为深红色油的标记化合物(11.75g)。
[0230]
(实施例3g)在将k2hpo4・
3h2o (108.4g)和kh2po
4 (37.82g)溶解于水(3780ml)而调制的缓冲液(3780ml)中加入kred (1279g)、d-葡萄糖(253g)、nadp (12.61g)、gdh (25.26g),制成混合溶液(ms-6-1),之后搅拌1小时。将进行与(实施例2a-2f)的方法同样的操作而得到的式(a-6)的化合物(126.05g)溶解于甲苯(630ml)而得到的混合溶液加入到先前调制的混合溶液(ms-6-1)中。在反应温度23℃ (20~25℃)下将反应溶液的ph保持在ph=6.0~7.0,同时搅拌26小时。在反应溶液中加入叔戊醇(500ml)和异戊醇(130ml),在反应温度23℃ (20~25℃)下搅拌16小时。加入乙酸乙酯(1300ml)、硅藻土(126g),升温至60℃,在同一温度下搅拌1小时。冷却至20℃后进行过滤,分离水层和有机层,将水层用乙酸乙酯(1300ml)进行萃取,与先前得到的有机层合并,之后用水(1300ml)进行洗涤,得到了有机层(op-6-1)。另外,在过滤后的硅藻土中加入乙酸乙酯(1300ml),在20~30℃下搅拌10小时,进行过滤,得到了有机层(op-6-2),再次于过滤后的硅藻土中加入乙酸乙酯(1300ml),在20~30℃下搅拌2小时,进行过滤,得到了有机层(op-6-3)。将有机层(op-6-1)、有机层(op-6-2)和有机层(op-6-3)合并,制成了有机层(op-6a)。此外,使用进行与(实施例2a-2f)的方法同样的操作而得到的式(a-6)的化合物(113g),进行与上述的方法同样的操作,进行反应,由此得到了有机层(op-6b)。将有机层(op-6a)和有机层(op-6b)合并后进行浓缩,由此得到了作为红褐色油的标记化合物(331g)。
[0231]
[式(a-7)的化合物的物性数据]
(1h nmr、400 mhz、生产商:bruker、cdcl3、δ ppm)关于式(a-7)的化合物的绝对立体构型的确定,在将式(a-7)的化合物转化成式(b)的化合物后,通过与按照国际公开第2003/095420号小册子等中记载的方法另外合成的式(b)的化合物的分析值对比并一致来进行。而且,将式(b)的化合物转化成其氢溴酸盐,将其氢溴酸盐用x射线晶体结构进行分析,由此确定了式(b)的化合物内的羟基为(r)构型(参照(实施例5)和图2)。
[0232]
(实施例4a) (r)-8-氨基-1,2,3,4-四氢萘-2-醇(式b-hcl)的盐酸盐合成[化学式99]在反应容器中装入正proh (2g),在-5~5℃下进行搅拌。在同一温度下用10分钟滴加乙酰氯(0.76g)。将反应温度升至50~55℃,之后用45分钟加入进行与(实施例3a-g)的方法同样的操作而得到的式(a-7)的化合物(0.5g)的正proh (6g)溶液。在反应温度50~55℃下搅拌45分钟,之后放置冷却使反应温度达到20~30℃之间,在20~30℃下搅拌16小时。过滤所得的固体,用正proh (5ml
×
2次)进行洗涤,在40~50℃下干燥6.5小时,由此得到了标记化合物(0.23g)。
[0233]
(实施例4b) (r)-8-氨基-1,2,3,4-四氢萘-2-醇(式b)的合成[化学式100]在反应容器中装入正proh (1.81g),在-5~5℃下进行搅拌。在同一温度下用10分钟滴加乙酰氯(2.35g)。将反应温度升至33~37℃,之后用45分钟加入进行与(实施例3a-g)
的方法同样的操作而得到的式(a-7)的化合物(4.25g)的正proh (12.75g)溶液。在反应温度33~37℃下搅拌49.5小时,之后在反应温度20~25℃下搅拌19小时。过滤所生成的固体,用i-proac (10ml
×
2次)进行洗涤,在30~40℃下干燥4小时,由此得到了盐酸盐(2.04g)。合并另外合成的盐酸盐(0.07g),达到2.11g,之后悬浮于乙酸乙酯(12ml)中,使用碳酸氢钠水溶液将水层的ph调节至7~8。分离水层和有机层,之后将水层用乙酸乙酯(12ml
×
2次)进行萃取,合并有机层,用水(10ml
×
2次)进行洗涤,在减压下浓缩有机层,由此得到了标记化合物(1.34g)。
[0234]
(实施例4c) (r)-8-氨基-1,2,3,4-四氢萘-2-醇(式b)的合成在反应容器中装入正proh (138g),在-5~5℃下进行搅拌。在同一温度下用1小时滴加乙酰氯(180.1g)。将反应温度升至33~37℃,之后用45分钟加入进行与(实施例3a-g)的方法同样的操作而得到的式(a-7)的粗化合物(331g)的正proh (750ml)溶液。在反应温度33~37℃下搅拌15小时,之后在反应温度50~55℃下搅拌2小时10分钟。在反应温度达到33~37℃后,在33~37℃下用30分钟加入将hcl (gass) (26g)加入到i-proac (192g)中而得的混合液。将反应温度升至50~55℃,搅拌1.5小时。过滤所生成的固体,用i-proac进行洗涤,将所得固体在减压下、20~30℃下干燥36小时,由此得到了盐酸盐(158g)。使该盐酸盐(158g)悬浮于乙酸乙酯(1000ml)中,用30分钟加入碳酸氢钠水溶液(76g的碳酸氢钠、1000ml的水)。分离水层和有机层,之后将水层用乙酸乙酯(1000ml
×
2次)进行萃取,合并有机层,用水(1000ml)进行洗涤,在减压下浓缩有机层,由此得到了标记化合物(136g)。
[0235]
[式(b)的化合物的物性数据](1h-nmr、400 mhz、生产商:bruker、cdcl3、δ ppm)(实施例5) (r)-8-氨基-1,2,3,4-四氢萘-2-醇氢溴酸盐的合成(式(b-hbr)):[化学式101]将以与(实施例4b)或(实施例4c)同样的操作而得到的(r)-8-氨基-1,2,3,4-四氢萘-2-醇的粗化合物(99.9g)溶解于乙酸乙酯(1l)中,在冰水冷下,加入48%氢溴酸水溶液(80ml)。过滤收集已沉淀的固体,用异丙醇(300~400ml)、乙酸乙酯(500ml)进行依次洗涤。将所得的粗氢溴酸盐(123.9g、98.3%ee)溶解于热水(250ml)中,加入活性炭(20g)。将活性炭趁热使用硅藻土进行过滤,并用水进行洗涤。减压浓缩滤液,将所得的残渣用水进行重结
晶。过滤收集所得到的晶体,用异丙醇、乙酸乙酯进行洗涤,得到了标记化合物(34.2g、98.4%ee)。收集滤液且进行减压浓缩,用水进行2次重结晶操作,得到了标记化合物(29.1g、98.9%ee)。
[0236]
式(b)的化合物的氢溴酸盐的光学纯度使用岛津hplc lc-10系统,按照以下的条件进行测定。
[0237]
[表13]另外,使用理学的afc-7来分析所得的式(b)的化合物((r)-8-氨基-1,2,3,4-四氢萘-2-醇)的氢溴酸盐的单晶的x射线晶体结构,得到了以下的结果(参照图2)。
[0238]
[表14](实施例6) (e)-2-(7-三氟甲基色满-4-亚基)-n-[(7r)-7-羟基-5,6,7,8-四氢萘-1-基]乙酰胺(式(i))的合成:[化学式102]
在(实施例5)中得到的(r)-8-氨基-1,2,3,4-四氢萘-2-醇氢溴酸盐(式(b-hbr)) (0.47g)与通过国际公开第2007/010383号小册子中记载的制造方法得到的(e)-2-(7-(三氟甲基)色满-4-亚基)乙酸(式(ca-1)) (0.50g、1.0eq)的甲醇(5.00ml:相对于1g的式(b-hbr)的化合物为约10容量倍)悬浮液中,加入三乙胺(0.27ml、1.0eq)和4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉盐酸盐(dmt-mm) (0.80g、1.5eq),在室温下搅拌3小时。将反应液进行冰冷后,过滤收集已沉淀的晶体,用冷甲醇进行洗涤。将所得的固体加热溶解于乙醇(6ml)后,在加热时加入水(6ml)。放置冷却后,过滤已沉淀的固体,用50%水-乙醇和水依次洗涤后,进行减压干燥,得到了作为白色固体的标记化合物(0.56g)。
[0239]
[式(i)的化合物的物性数据](1h-nmr数据(cdcl3) (δ:ppm)):(lc-ms):rt=4.73 (分钟)、[m+h]
+
=404光学纯度:97.9%ee。
[0240]
式(i)的化合物的光学纯度使用岛津hplc lc-vp系统,按照以下的条件进行测定。
[0241]
[表15]使用理学的r-axis v检测器在spring-8光束线bl32b2处分析式(i)的化合物的晶体结构(参照图3)。
[0242]
[表16]
(实施例7) (e)-2-(7-三氟甲基色满-4-亚基)-n-[(7r)-7-羟基-5,6,7,8-四氢萘-1-基]乙酰胺的合成:(缩合反应的条件研究(1))[化学式103]《研究1》:在(实施例5)中得到的(r)-8-氨基-1,2,3,4-四氢萘-2-醇氢溴酸盐(200mg)与通过国际公开第2007/010383号小册子中记载的制造方法得到的(e)-2-(7-(三氟甲基)色满-4-亚基)乙酸(212mg、1.0eq)的甲醇(3.15ml:相对于1g的式(b-hbr)的化合物为15.75容量倍)悬浮液中,加入三乙胺(120μl、1.05eq)和4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉盐酸盐(dmt-mm) (339mg、1.5eq),在室温下搅拌3小时。加入水,过滤已沉淀的固体,用水洗涤后,进行干燥,得到了作为白色固体的标记化合物(291mg)。
[0243]
《研究2》:在(实施例5)中得到的(r)-8-氨基-1,2,3,4-四氢萘-2-醇氢溴酸盐(200mg)与通过国际公开第2007/010383号小册子中记载的制造方法得到的(e)-2-(7-(三氟甲基)色满-4-亚基)乙酸(212mg、1.0eq)的甲醇(3.15ml:相对于1g的式(b-hbr)的化合物为15.75容量倍)悬浮液中,加入三乙胺(240μl、2.1eq)和4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉盐酸盐(dmt-mm) (339mg、1.5eq),在室温下搅拌3小时。加入水,过滤已沉淀的固体,用水洗涤后,进行干燥,得到了作为白色固体的标记化合物(273mg)。
[0244]
《研究3》:在(实施例5)中得到的(r)-8-氨基-1,2,3,4-四氢萘-2-醇氢溴酸盐(200mg)与通过国际公开第2007/010383号小册子中记载的制造方法得到的(e)-2-(7-(三
氟甲基)色满-4-亚基)乙酸(212mg、1.0eq)的甲醇(1.60ml:相对于1g的式(b-hbr)的化合物为8容量倍)悬浮液中,加入三乙胺(120μl、1.05eq)和4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉盐酸盐(dmt-mm) (339mg、1.5eq),在室温下搅拌3小时。加入水,过滤已沉淀的固体,用水洗涤后,进行干燥,得到了作为白色固体的标记化合物(298mg)。
[0245]
《研究4》:在(实施例5)中得到的(r)-8-氨基-1,2,3,4-四氢萘-2-醇氢溴酸盐(200mg)与通过国际公开第2007/010383号小册子中记载的制造方法得到的(e)-2-(7-(三氟甲基)色满-4-亚基)乙酸(212mg、1.0eq)的甲醇(3.15ml:相对于1g的式(b-hbr)的化合物为15.75容量倍)悬浮液中,加入三乙胺(120μl、1.05eq)和4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉盐酸盐(dmt-mm) (339mg、1.5eq),在加热回流下搅拌3小时。放置冷却后,加入水,过滤已沉淀的固体,用水洗涤后,进行干燥,得到了作为白色固体的标记化合物(291mg)。
[0246]
在上述(实施例7)的《研究1》~《研究4》中,式(i)的化合物的endo型((r)-n-(7-羟基-5,6,7,8-四氢萘-1-基)-2-(7-(三氟甲基)-2h-色满-4-亚基)乙酰胺)的生成得到抑制。
[0247]
式(i)的化合物的化学纯度使用岛津hplc lc-vp系统,按照以下的条件进行测定。
[0248]
[表17](实施例8) (e)-2-(7-三氟甲基色满-4-亚基)-n-[(7r)-7-羟基-5,6,7,8-四氢萘-1-基]乙酰胺的合成:(缩合反应的条件研究(2))[化学式104]《研究1》:在以与(实施例4b)或(实施例4c)同样的操作而得到的(r)-8-氨基-1,2,3,4-四氢萘-2-醇(101mg)和通过国际公开第2007/010383号小册子中记载的制造方法得到的(e)-2-(7-(三氟甲基)色满-4-亚基)乙酸(200mg、1.25eq)的异丙醇(3.0ml:相对于1g的式(b)的化合物为约30容量倍)悬浮液中,加入4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉盐酸盐(dmt-mm) (257mg、1.5eq),在室温下搅拌6小时。加入水(3ml),过滤已沉淀的固体,用水洗涤后,进行干燥,得到了作为白色固体的标记化合物(209mg)。
[0249]
《研究2》:在以与(实施例4b)或(实施例4c)同样的操作而得到的(r)-8-氨基-1,2,3,4-四氢萘-2-醇(126mg)和通过国际公开第2007/010383号小册子中记载的制造方法得到的(e)-2-(7-(三氟甲基)色满-4-亚基)乙酸(200mg、1.0eq)的异丙醇(3.00ml:相对于1g的式(b)为约24容量倍)悬浮液中,加入4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉盐酸盐(dmt-mm) (257mg、1.5eq),在室温下搅拌6小时。加入水(3ml),过滤已沉淀的固体,用水洗涤后,进行干燥,得到了作为白色固体的标记化合物(238mg)。
[0250]
《研究3》:在以与(实施例4b)或(实施例4c)同样的操作而得到的(r)-8-氨基-1,2,3,4-四氢萘-2-醇(152mg)和通过国际公开第2007/010383号小册子中记载的制造方法得到的(e)-2-(7-(三氟甲基)色满-4-亚基)乙酸(200mg、0.83eq)的异丙醇(3.00ml:相对于1g的式(b)的化合物为约20容量倍)悬浮液中,加入4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉盐酸盐(dmt-mm) (257mg、1.5eq),在室温下搅拌6小时。加入水(3ml),过滤已沉淀的固体,用水洗涤后,进行干燥,得到了作为白色固体的标记化合物(243mg)。
[0251]
《研究4》:在以与(实施例4b)或(实施例4c)同样的操作而得到的(r)-8-氨基-1,2,3,4-四氢萘-2-醇(177mg)和通过国际公开第2007/010383号小册子中记载的制造方法得到的(e)-2-(7-(三氟甲基)色满-4-亚基)乙酸(200mg、0.71eq)的异丙醇(3.00ml:相对于1g的式(b)的化合物为约20容量倍)悬浮液中,加入4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉盐酸盐(dmt-mm) (257mg、1.5eq),在室温下搅拌6小时。加入水(3ml),过滤已沉淀的固体,用水洗涤后,进行干燥,得到了作为白色固体的标记化合物(238mg)。
[0252]
(实施例9a) (e)-2-(7-三氟甲基色满-4-亚基)-n-[(7r)-7-羟基-5,6,7,8-四氢萘-1-基]乙酰胺的合成:使用以与(实施例4b)或(实施例4c)同样的操作而得到的(r)-8-氨基-1,2,3,4-四氢萘-2-醇(1.04g、纯度99.35%)、通过国际公开第2007/010383号小册子中记载的制造方法得到的(e)-2-(7-(三氟甲基)色满-4-亚基)乙酸(1.05eq)、异丙醇(相对于式(b)的化合物为25容量倍)和4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉盐酸盐(dmt-mm) (1.3eq)进行缩合反应(反应条件:20~25℃、搅拌22小时)后,依据(实施例8)进行后处理,由此得到了作为固体的标记化合物(1.87g)。
[0253]
(实施例9b) (e)-2-(7-三氟甲基色满-4-亚基)-n-[(7r)-7-羟基-5,6,7,8-四氢萘-1-基]乙酰胺的合成(扩大规模):使用以与(实施例4b)或(实施例4c)同样的操作而得到的(r)-8-氨基-1,2,3,4-四氢萘-2-醇(110g)、通过国际公开第2007/010383号小册子中记载的制造方法得到的(e)-2-(7-(三氟甲基)色满-4-亚基)乙酸(173.9g、1.05eq)、异丙醇(2600ml:相对于式(b)的化合物为25容量倍)和4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉盐酸盐(dmt-mm) (243.7g、1.3eq)进行缩合反应(反应条件:20~25℃、搅拌22小时)后,依据(实施例8)进行后处理,由此得到了作为灰白色固体的标记化合物(183.3g)。
[0254]
式(i)的化合物的光学纯度使用岛津hplc lc-vp系统,按照以下的条件进行测定。
[0255]
[表18]
在式(a8-br)和式(b)的化合物的核磁共振光谱(nmr)的测定中,使用了bruker av400。
[0256]
式(a8-br)和式(b)的高效液相色谱(hplc)按照以下的方法进行测定。
[0257]
[表19][表20]
[表21][表22]
[表23][表24][表25]
[表26](实施例10a)~(实施例10d) (r)-8-溴-1,2,3,4-四氢萘-2-醇(a8-br)的合成[化学式105](实施例10a)在配备有定轨振荡器(orbital shaker) (上海南荣实验室设备有限公司制、型号:nry-200)的烧瓶中混合kred (来自escherichia coli sp.的酮还原酶、5mg)、d-葡萄糖(200mg)、葡萄糖脱氢酶(gdh) (2mg)、烟酰胺腺嘌呤二核苷酸磷酸(nadp) (1mg)和磷酸缓冲液(3ml:将10.62g的kh2po4、21.25g的k2hpo4加入到1000ml水中而调制的缓冲液)进行搅拌,调制了混合溶液。其次,在先前调制的混合溶液中加入将8-溴-3,4-二氢萘-2(1h)-酮(式(sm8-br)) (100mg)溶解于二甲基亚砜(dmso) (0.3ml)而得的混合溶液,在反应温度30
2(1h)-酮(式(sm8-br)) (110.31g)溶解于二甲基亚砜(dmso) (330ml)而得的混合溶液。在反应温度20~25℃下搅拌1小时,之后使用2m碳酸钠水溶液使反应溶液的ph调节为ph=6.5~7.0的范围。接着,在反应温度20~25℃下搅拌1小时,之后使用2m碳酸钠水溶液使反应溶液的ph调节为ph=6.5~7.0的范围。再在反应温度20~25℃下搅拌2小时,之后使用2m碳酸钠水溶液使反应溶液的ph调节为ph=6.5~7.0的范围。其后,在反应温度20~25℃下搅拌16小时。取出一部分反应溶液进行hplc分析,由此确认到标记化合物的ipc收率为97.4%。
[0262]
在反应溶液中加入mtbe (1100ml),再加入包含水(110g)的硅藻土(diatomite) (110g),将混合溶液的温度设为50~60℃并搅拌30分钟。将混合溶液的温度放置冷却至20~25℃,再在同一温度下搅拌2小时。过滤上述混合溶液,将过滤物(湿滤饼,wet cake)用mtbe (110ml)进行洗涤,得到了滤液c。将上述湿滤饼装入到反应容器中,加入mtbe (900ml),在20~25℃下搅拌12小时。过滤包含湿滤饼的悬浮液,将湿滤饼用mtbe (110ml)进行洗涤,得到了滤液d。将滤液c和滤液d混合,分离水层和有机层,将水层用mtbe (1000ml)进行萃取,与先前得到的有机层合并,之后用水(675ml)进行洗涤,将有机层进行浓缩,由此得到了粗标记化合物(108.03g,光学纯度为99.8%)。
[0263]
[式(a8)的物性数据](1h nmr、400 mhz、生产商:bruker、dmso-d6、δ ppm)(实施例10a)~(实施例10d)中使用的kred (来自escherichia coli sp.的酮还原酶)是enzymeworks公司制造(产品编号:hq-k-105)的酶。
[0264]
(实施例10a)~(实施例10d)中得到的式(a8-br)的化合物的绝对立体构型是在将式(b)转化成式(a8-br)后,通过使其与国际公开第2003/095420号小册子等中记载的方法另外合成的式(b)的化合物的分析值一致来确定。
[0265]
(参考例1) 8-溴-1,2,3,4-四氢萘-2-醇(式(a8-br-rac)的合成[化学式106]在反应容器中加入8-溴-3,4-二氢萘-2(1h)-酮(式(sm8-br)) (20.0g)和甲醇(200ml),在内温为0~5℃下加入nabh
4 (8.28g),在同一温度下搅拌1小时(取出一部分反应溶液进行hplc分析,确认到ipc收率为98.5%)。在反应溶液的温度为5℃以下滴加10%碳酸
氢钠水溶液(1.5l),在混合溶液的温度为0~5℃下搅拌0.2小时。加入乙酸乙酯(1.5l),分离水层和有机层,将水层用乙酸乙酯(1.5l)进行萃取,与先前得到的有机层合并,之后用25wt%氯化钠水溶液(1.5l)进行洗涤,将有机层进行浓缩,由此得到了粗标记化合物(21.53g)。将所得的粗标记化合物进行硅胶柱色谱(正庚烷:乙酸乙酯=1:1),由此得到了标记化合物(21.29g)。确认到所得的式(a8-br-rac)的化合物与文献已知的物性数据一致。
[0266]
(实施例11a)~(实施例11g) (r)-8-氨基-1,2,3,4-四氢萘-2-醇(式(b))的合成[化学式107](实施例11a)进行与实施例10a~实施例10d的方法同样的操作,将在酶还原中得到的(r)-8-溴-1,2,3,4-四氢萘-2-醇(式(a8-br)) (100mg)、cu2o (40mg)、n-甲基吡咯烷酮(nmp) (2ml)和氨水(3ml)在封管反应容器中混合,在105~115℃的温度下进行20小时的封管反应。用水进行稀释,之后用乙酸乙酯进行萃取,将有机层用25wt%氯化钠水溶液进行洗涤,将有机层用na2so4进行干燥、过滤,之后将有机层进行浓缩,由此得到了粗标记化合物(106mg)。进行薄层色谱法(正庚烷:乙酸乙酯=1:1),并进行分离,由此得到了标记化合物(10mg) (光学纯度为96.8%)。
[0267]
(实施例11b)进行与实施例10a~实施例10d的方法同样的操作,将在酶还原中得到的(r)-8-溴-1,2,3,4-四氢萘-2-醇(式(a8-br)) (2.2g)、cu2o (700mg)、nmp (3.5ml、1.6v)和氨水(5.5ml)在封管反应容器中混合,在105~115℃的温度下进行37小时的封管反应(确认到ipc收率在16小时后为83.75%、在21小时后为88.91%、在37小时后为93.12%)。用水(17ml)和乙酸乙酯(11ml)进行稀释,之后进行过滤,将过滤物用乙酸乙酯(4ml、3次)进行洗涤,分离水层和有机层。然后,将水层用乙酸乙酯(11ml、5次)进行萃取,与先前得到的有机层合并,之后用水(20ml、2次)进行洗涤,将有机层用10%na2so4水溶液进行洗涤,将有机层进行浓缩,由此得到了粗标记化合物(1.25g、61.27%、光学纯度为95.6%)。
[0268]
(实施例11c)按照下述表中所示的条件进行封管反应,进行了反应溶剂的验证。
[0269]
[表27]
溴-1,2,3,4-四氢萘-2-醇(式(a8-br)) (16.32g、nmp溶液、含量61.1%)、cu2o (3.18g、0.51eq)、nmp (5ml、0.5v)和氨水(35ml、3.5v)在封管反应容器中混合,在105~115℃的温度下进行40小时的封管反应(确认到第40小时的ipc收率为90.16%)。将反应溶液冷却后,在反应溶液中加入25wt%氯化钠水溶液(75ml)和2-methf (50ml),使用硅藻土(diatomite) (20.00g)进行过滤,将过滤物(滤饼)用2-methf (50ml)进行洗涤。分离水层和有机层,将水层用2-methf (50ml、2次)进行萃取,与先前得到的有机层合并,之后用8wt%na2so4水溶液(50ml、2次)进行洗涤,取出有机层(总量的92.5%的量),在5~15℃下滴加0.5m盐酸水溶液(111ml) (此时的ph=1.2)。分离水层和有机层,将水层用2-methf (30ml)进行萃取。在水层中加入10%氢氧化钠水溶液(22ml),用2-methf (100ml、50ml、50ml)进行萃取。与先前得到的有机层合并,之后在40℃以下浓缩有机层,在35~45℃下滴加正庚烷(80ml),之后冷却至5℃。其后,在0~10℃下搅拌24小时,进行过滤收集,将过滤物用正庚烷(10ml)进行洗涤并干燥,由此得到了标记化合物(4.50g、67.8%、光学纯度为99.9%)。
[0272]
(实施例11g)按照下述表中的条件进行了封管反应。
[0273]
[表29](后处理11g-1)结束实施例11g-1的反应,将反应溶液冷却后,在反应溶液中加入25wt%氯化钠水溶液(345ml)和2-methf (250ml),使用硅藻土进行过滤,将过滤物(滤饼)用2-methf (230ml)进行洗涤。分离水层和有机层,将水层用2-methf (230ml、2次)进行萃取,得到了与先前得到的有机层合并的有机相(11g-1)。
[0274]
(后处理11g-2)结束实施例11g-2的反应,将反应溶液冷却后,在反应溶液中加入25wt%氯化钠水溶液(375ml)和2-methf (250ml),使用硅藻土进行过滤,将过滤物(滤饼)用2-methf (250ml)进行洗涤。分离水层和有机层,将水层用2-methf (250ml、2次)进行萃取,得到了与先前得到的有机层合并的有机相(11g-2)。
[0275]
(后处理11g-3)将先前得到的有机相(11g-1)和有机相(11g-2)混合,之后用8wt%na2so4水溶液
(480ml、2次)进行洗涤,之后滴加0.5m盐酸水溶液(1156ml),调节为ph=0.88。分离水层和有机层,将水层用2-methf (290ml)进行萃取。在水层中加入10%氢氧化钠水溶液(230ml),用2-methf (1000ml、500ml、500ml、3次)进行萃取。与先前得到的有机层合并,之后在40℃以下浓缩有机层,在35~45℃下滴加正庚烷(576ml),之后冷却至0~10℃,在同一温度下进行搅拌,之后进行过滤收集,将过滤物用正庚烷(96ml)进行洗涤并干燥,由此得到了标记化合物(53.55g、70.8%、光学纯度为99.9%)。
[0276]
[式(b)的物性数据](1h-nmr、400 mhz、生产商:bruker、cdcl3、δ ppm)(实施例11a)~(实施例11g)中使用的氨水是25~28%的氨水。
[0277]
(实施例12a)~(实施例12b) (e)-2-(7-三氟甲基色满-4-亚基)-n-[(7r)-7-羟基-5,6,7,8-四氢萘-1-基]乙酰胺的合成[化学式108](实施例12a)使用以与(实施例11g)同样的操作而得到的(r)-8-氨基-1,2,3,4-四氢萘-2-醇(3g)、通过国际公开第2007/010383号小册子中记载的制造方法得到的(e)-2-(7-(三氟甲基)色满-4-亚基)乙酸(4.83g、1.0eq)、异丙醇(60.01g:相对于式(b)的化合物为20重量倍)和4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉盐酸盐(dmt-mm) (6.67g、1.3eq)进行了缩合反应(反应条件:20~25℃、搅拌17小时)。在反应溶液中加入水(75ml),之后冷却至10~15℃,在同一温度下搅拌1小时,之后进行过滤收集,将过滤物用水进行洗涤,将过滤物在36℃下干燥16小时。在所得的过滤物中加入2-methf (90g),在70~80℃下搅拌0.5小时,之后将溶液在40℃以下进行减压浓缩直至溶液的容量达到6ml。接着,将所浓缩的溶液在70~80℃下搅拌1小时,之后冷却至0~5℃,在同一温度下滴加60g正庚烷,再在同一温度下搅拌30分钟。进行过滤收集,将过滤物用正庚烷(9g)进行洗涤,将过滤物进行干燥,由此得到了标记化合物(5.96g、光学纯度为99.9%)。
[0278]
(实施例12b) [扩大规模]:使用以与(实施例11g)同样的操作而得到的(r)-8-氨基-1,2,3,4-四氢萘-2-醇(44.5g)、通过国际公开第2007/010383号小册子中记载的制造方法得到的(e)-2-(7-(三氟甲基)色满-4-亚基)乙酸(72.5g、1.0eq)、异丙醇(900g:相对于式(b)的化合物为18重量倍)
和4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉盐酸盐(dmt-mm) (100.05g、1.3eq)进行了缩合反应(反应条件:20~25℃、搅拌13小时)。通过依据(实施例12a)进行后处理,得到了标记化合物(95.7g、光学纯度为100%)。
[0279]
[式(i)的化合物的物性数据](1h-nmr数据(cdcl3) (δ:ppm)):(lc-ms):rt=4.73 (分钟)、[m+h]
+
=404。
[0280]
式(i)的化合物的光学纯度使用岛津hplc lc-vp系统,按照以下的条件进行测定。
[0281]
[表30]符号说明l1、l2、l3、l4:氮导入口;m1:包含原料+tempo+二氯甲烷的容器;m2:包含kbr+nahco3+水的容器;m3:包含naclo的容器;p1、p2、p3:泵;t1、t2、t3:预冷管;s1、s2、s3:搅拌机;r1、r2、r3:反应容器;cd:回收桶。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1