噻吩类化合物及其制备与作为肠道病毒抑制剂的应用的制作方法
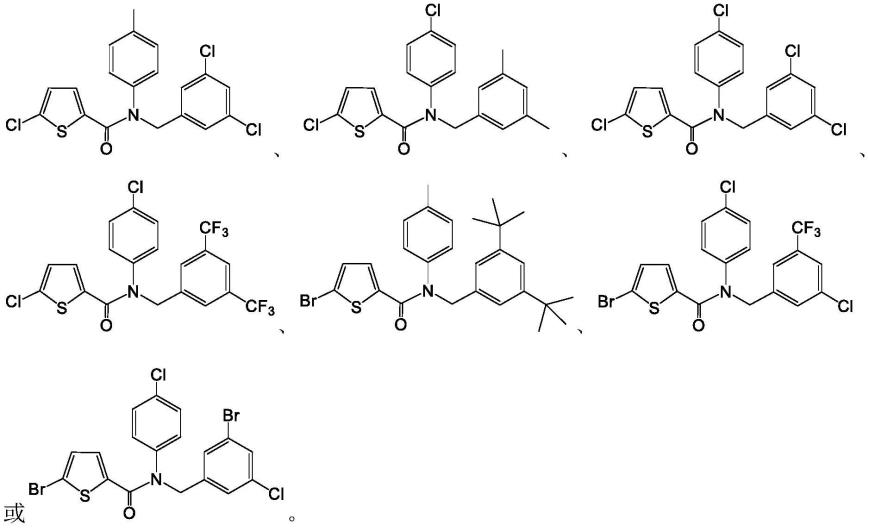
1.本发明属于药物化学领域,具体涉及一种噻吩类化合物及其制备与作为肠道病毒抑制剂的应用。
背景技术:
2.肠道病毒(enteroviruses,简写为ev),是一类侵入肠细胞并在其中繁殖的病原体,可诱导人肠上皮细胞产生多种炎症。1970年,国际病毒命名委员会将其归属于微小核糖核酸病毒科的肠道病毒属,按照肠道病毒序数编号命名,即68﹑69﹑70﹑71﹑72型肠道病毒等。
3.其中,
4.肠道病毒71型(ev71),是无包膜的单链rna病毒,于1969年首次从患有脑炎的婴儿的粪便中分离出来,是手足口病的主要致病因子。
5.该病毒是以引起婴幼儿以发热和手、足、口腔等部位的皮疹、疱疹为主要特征的急性传染病。此外,该病毒还可能导致严重的神经系统疾病,例如:脑炎、脑膜炎等,对新生儿的生命健康造成严重威胁。
6.ev71感染是一种全球性的传染病,世界大部分地区均有此病爆发和流行的相关报道,受到世界各国广泛的关注和警惕。然而,不幸的是,目前仍然缺乏能够有效治疗手足口病的药物。因此,设计、合成一系列新型有效的ev71抑制剂就显得尤为迫切。
7.有鉴于此,特提出本发明。
技术实现要素:
8.针对现有技术存在的问题和/或不足,本发明的目的在于提供一种噻吩类化合物及其制备与作为肠道病毒抑制剂的应用。该类化合物,是将噻吩通过酰胺基团与对位取代的苯基以及3,5位取代的苄基相连接,结构新颖独特,对ev71、evd68等多种肠道病毒具有良好的抑制活性以及较小的细胞毒性,为药学上肠道病毒感染用药提供了一种新的潜在选择。
9.本发明提供的一种式ⅰ所示的化合物或其药学上可接受的盐、晶型、溶剂合物;
[0010][0011]
其中,r1、r2、r3、r4独立地选自卤素、c1~c4烷基或卤素取代的c1~c4烷基。
[0012]
进一步的,
[0013]
在上述任一技术方案(化合物或其盐、晶型、溶剂合物)中,r1为卤素;r2为卤素或c1~c4烷基;r3、r4独立地选自卤素、c1~c4烷基或卤素取代的c1~c4烷基;优选的,所述的卤素
选自氟、氯或溴,和/或,所述的c1~c4烷基选自甲基、乙基、正丙基、异丙基、正丁基、异丁基或叔丁基。
[0014]
进一步的,
[0015]
在上述任一技术方案(化合物或其盐、晶型、溶剂合物)中,r1为氟、氯或溴;r2为氯或甲基;r3、r4独立地选自氟、氯、溴、甲基、叔丁基、氟取代的甲基或氟取代的叔丁基。
[0016]
进一步的,
[0017]
在上述任一技术方案(化合物或其盐、晶型、溶剂合物)中,式ⅰ所示的化合物为:
[0018]
本发明还提供了式ⅰ所示化合物的制备方法,它包括以下步骤:
[0019]
i、
[0020][0021]
在卤代烃类溶剂中以及有机胺存在的条件下,化合物5与化合物4发生缩合脱去hcl的反应,生成式ⅰ所示的化合物;
[0022]
ii、
[0023][0024]
化合物1与化合物2在醇类溶剂中发生缩合脱去h2o的反应,生成化合物3,再经mbh4还原,生成化合物4;
[0025]
其中,r1、r2、r3、r4的定义如前面任意一项所述,m为碱金属。
[0026]
进一步的,
[0027]
上述任一技术方案(式ⅰ所示化合物的制备方法),步骤i中,
[0028]
化合物4与化合物5之间的摩尔比为1:0.9~1.5,优选为1:1.0~1.2;
[0029]
和/或,
[0030]
每克化合物4对应的醇类溶剂用量为10~80ml,优选为45~60ml;
[0031]
和/或,
[0032]
化合物4与有机胺之间的摩尔比为1:0.9~1.5,优选为1:1.0~1.4;
[0033]
和/或,
[0034]
缩合脱去hcl的反应温度为10~40℃,优选为10~30℃;
[0035]
和/或,
[0036]
缩合脱去hcl的反应时间为3~24h,优选为5~12h。
[0037]
进一步的,
[0038]
上述任一技术方案(式ⅰ所示化合物的制备方法),步骤ii中,
[0039]
化合物1与化合物2之间的摩尔比为1:0.9~1.5,优选为1:0.9~1.2;
[0040]
和/或,
[0041]
每克化合物1对应的醇类溶剂用量为10~80ml,优选为45~60ml;
[0042]
和/或,
[0043]
化合物1与mbh4之间的摩尔比为1:0.9~1.5,优选为1:1.2~1.5;
[0044]
和/或,
[0045]
缩合脱去h2o的反应温度为10~40℃,优选为10~30℃;
[0046]
和/或,
[0047]
缩合脱去h2o的反应时间为3~24h,优选为5~20h;
[0048]
和/或,
[0049]
mbh4还原的反应温度为10~40℃,优选为10~30℃;
[0050]
和/或,
[0051]
mbh4还原的反应时间为1~12h,优选为2.5~5h。
[0052]
进一步的,
[0053]
在上述任一技术方案(式ⅰ所示化合物的制备方法)中,
[0054]
所述的卤代烃类溶剂为二氯甲烷;
[0055]
和/或,
[0056]
所述的有机胺为三乙胺;
[0057]
和/或,
[0058]
所述的醇类溶剂为甲醇或乙醇;
[0059]
和/或,
[0060]
所述的mbh4为硼氢化钠或硼氢化钾。
[0061]
此外,
[0062]
本发明还提供了前面所述式ⅰ所示化合物或其药学上可接受的盐、晶型、溶剂合物在制备肠道病毒抑制剂中的应用;例如,所述的肠道病毒是ev71、evd68、cva16、cvb1或
echov-6。
[0063]
本发明还提供了一种治疗和/或预防肠道病毒感染疾病的药物组合物:
[0064]
它是以前面任意一项所述式ⅰ所示化合物或其药学上可接受的盐、晶型、溶剂合物作为活性成分,加上药学上常用的辅料制成的制剂;例如,所述的肠道病毒感染疾病是手足口病。
[0065]
本发明的化合物,具有基本核心部分:将噻吩通过酰胺基团与对位取代的苯基以及3,5位取代的苄基相连接,结构新颖独特。体外细胞试验表明,本发明化合物对ev71、evd68、cva16、cvb1、echov-6等肠道病毒,具有良好的抑制活性,ec
50
在0.1~5.0μm之间,以及较小的细胞毒性,cc
50
大于50μm。体内生物活性试验表明,本发明化合物能够有效提高肠道病毒感染小鼠的存活率,进一步证明了化合物对肠道病毒的抑制作用和/或显著疗效,为药学上肠道病毒感染用药提供了一种新的潜在选择。
[0066]
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
具体实施方式
[0067]
下面将结合具体实施例对本发明进行清楚、完整的描述,本领域技术人员将会理解,下面所述的实施例是本发明一部分实施例,而不是全部的实施例,仅用于说明本发明,而不应视为对本发明保护范围的限制。
[0068]
本发明中,未注明具体条件者,按照常规条件或制造商建议的条件进行,所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0069]
关于本发明中使用术语的定义,除非另有说明,本文中术语提供的初始定义适用于全文中的该术语;对于本文没有具体定义的术语,应当根据公开内容和/或上下文,给出本领域技术人员能够给予它们的含义。
[0070]
ca~cb烷基,表示任何包含a~b个碳原子的烷基,例如:c1~c3烷基是指包含1~3个碳原子的烷基,例如:甲基、乙基、丙基等。
[0071]
术语“药学上可接受的”,通常在化学上或物理上与构成某药物剂型的其它成分相兼容,并在生理上与受体相兼容。
[0072]
术语“盐”,是指上述化合物或其异构体,与无机和/或有机的酸、碱形成酸式和/或碱式盐,也包括两性离子盐,还包括季铵盐,例如,烷基铵盐等。
[0073]
术语“溶剂合物”,是指一个或多个溶剂分子与本发明化合物形成缔合物。形成溶剂合物的溶剂包括但并不限于:水、甲醇、乙醇、氨基乙醇、异丙醇、二甲基亚砜、乙酸、乙酸乙酯等。
[0074]
实施例1
[0075]
1、化合物b的制备
[0076]
[0077]
将12g(约0.11mol)对甲基苯胺和19.5g(约0.11mol)3,5-二氯苯甲醛,加入到650ml乙醇中,室温搅拌反应15h(缩合反应),生成化合物a(中间产物),不经分离,之后分批或者逐步加入5.8g(约0.15mol)硼氢化钠,继续室温搅拌反应3h(还原反应),生成化合物b,浓缩,析出固体,抽滤,用乙醇或乙醇-石油醚(体积比2:1)重结晶,干燥,得到化合物b,白色或浅黄色固体,收率65.8%。
[0078]
化合物b:c
14h13
cl2n,m/z:265.0422;熔点:176~178℃。
[0079]
化合物b的核磁数据:1h nmr(dmso-d6):δ7.58(s,h),7.35(s,h),7.31(d,2h),7.09(d,2h),6.47(d,2h),4.32(d,2h),2.32(m,3h);
13
c nmr(dmso-d6):δ146.3,144.4,135.5,129.6,128.4,127.2,113.4,47.0,21.3。
[0080]
2、化合物c的制备
[0081][0082]
将20g(约0.11mol)5-氯-2-噻吩甲酰氯、27g(约0.1mol)化合物b和12.5g(约0.12mol)三乙胺,加入到1500ml二氯甲烷中,15℃下搅拌反应8h,hplc或tlc监测反应完全后,浓缩,过硅胶柱纯化(流动相:石油醚-乙酸乙酯=1:2,体积比),得到化合物c,白色或浅黄色固体,收率45.3%。
[0083]
化合物c:c
19h14
cl3nos,m/z:408.9857。
[0084]
化合物c的核磁数据:1h nmr(dmso-d6):δ8.08(s,h),7.60(s,h),7.32(m,4h),7.14(d,2h),5.40(d,2h),2.31(m,3h);
13
c nmr(dmso-d6):δ163.7,144.4,140.7,138.3,136.8,136.1,135.9,135.5,133.4,129.8,129.2,128.4,127.2,48.4,21.3。
[0085]
实施例2
[0086]
1、化合物d的制备
[0087][0088]
以对氯苯胺和3,5-二甲基苯甲醛为原料,按照与实施例1类似的方法,制备得到化合物d,收率64.5%。化合物d:c
15h16
cln,m/z:245.0965。
[0089]
2、化合物e的制备
[0090][0091]
以5-氯-2-噻吩甲酰氯和化合物d为原料,按照与实施例1类似的方法,制备得到化合物e,收率39.8%。化合物e:c
20h17
cl2nos,m/z:389.0406。
[0092]
实施例3
[0093][0094]
按照与实施例1类似的方法,以对氯苯胺和3,5-二氯苯甲醛为原料,制备得到化合物f;然后,再以5-氯-2-噻吩甲酰氯和化合物f为原料,制备得到化合物g。
[0095]
实施例4
[0096][0097]
按照与实施例1类似的方法,以对氯苯胺和3,5-双(三氟甲基)苯甲醛为原料,制备得到化合物h;然后,再以5-氯-2-噻吩甲酰氯和化合物h为原料,制备得到化合物i。
[0098]
实施例5
[0099][0100]
按照与实施例1类似的方法,以对甲基苯胺和3,5-双(叔丁基)苯甲醛为原料,制备得到化合物j;然后,再以5-溴-2-噻吩甲酰氯和化合物j为原料,制备得到化合物k。
[0101]
实施例6
[0102]
[0103]
按照与实施例1类似的方法,以对氯苯胺和3-氯-5-(三氟甲基)苯甲醛为原料,制备得到化合物l;然后,再以5-溴-2-噻吩甲酰氯和化合物l为原料,制备得到化合物m。
[0104]
实施例7
[0105][0106]
按照与实施例1类似的方法,以对氯苯胺和3-溴-5-氯苯甲醛为原料,制备得到化合物n;然后,再以5-溴-2-噻吩甲酰氯和化合物n为原料,制备得到化合物o。
[0107]
实施例8
[0108]
1、细胞毒性试验
[0109]
cell counting kit-8,简称cck-8试剂盒,是一种广泛应用于细胞增殖和细胞毒性的快速高灵敏度检测试剂盒。其工作原理是:在电子耦合试剂存在的情况下,被线粒体内的脱氢酶还原生成高度水溶性的橙黄色的甲臜产物。颜色的深浅,与细胞的增殖成正比,与细胞毒性成反比。使用酶标仪测定od值,间接反映活细胞数量。
[0110]
试验时,向铺满rd细胞的96孔板中,24小时后加入不同稀释度的药物。37℃恒温条件下培养8~24小时后,加入20μl mts/pms混合溶液,37℃恒温条件下继续培养4小时,用酶标仪测定450nm下的od值。
[0111]
化合物的抑制率(%)=[1-(e-n)/(p-n)]
×
100,其中,“e”代表实验组的od值,“p”代表阳性对照组的od值,“n”代表阴性对照组的od值。用cc
50
(half-cytotoxic concentration)作为化合物细胞毒性的评价指标。
[0112]
2、抗ev71活性试验
[0113]
ev71病毒感染vero、rd等细胞后,经过一定的时间会引起细胞病变,可以根据细胞病变的程度来反映药用化合物对病毒的抑制水平。
[0114]
试验时,向铺满rd细胞的96孔板中加入ev71病毒与不同稀释度的药物的混合物,面积比为80%,每个稀释度8个重复孔。37℃恒温条件下培养24~48小时,病毒对照组细胞完全病变后,加入20μl mts/pms混合溶液,37℃恒温条件下继续培养4小时,用酶标仪测定490nm下的od值。
[0115]
化合物的抑制率(%)=[1-(e-n)/(p-n)]
×
100,其中,“e”代表实验组的od值,“p”代表阳性对照组的od值,“n”代表阴性对照组的od值。用ec
50
(half-maximal effective concentration)作为抗病毒活性的评价指标。
[0116]
以恩韦肟(enviroxime)作为对照,分别对前述实施例制备的化合物c、化合物e、化合物g、化合物i、化合物k、化合物m、化合物o进行细胞毒性试验、抗ev71活性试验,并计算化合物的选择性指数si(selectivity index),结果见表1。
[0117]
cc
50
:半数细胞毒性的浓度,数值越高说明对细胞的毒性越低;
[0118]
ec
50
:半数有效浓度,数值越小说明对病毒的抑制效果越好;
[0119]
si:为cc
50
与ec
50
的比值,数值越大说明成药的安全性和/或可能性越高。
[0120]
表1、本发明合成化合物的细胞毒性和抗ev71活性结果
[0121]
试验对象cc
50
(μm)ec
50
(μm)si化合物c158.4700.204776.8化合物e134.1260.225596.1化合物g125.3950.308407.1化合物i147.8020.257575.1化合物k160.2280.1201335.2化合物m152.3040.262581.3化合物o130.4960.285457.9恩韦肟28.00.140200
[0122]
此外,
[0123]
研究还发现,本发明的前述化合物,对evd68、cva16、cvb1、echov-6等肠道病毒也具有良好的抑制活性,ec
50
在0.1~5.0μm之间,cc
50
大于50μm。
[0124]
实施例9
[0125]
体内生物活性测试:
[0126]
试验分组:空白对照组、病毒对照组、化合物c处理组、化合物e处理组、化合物k处理组和恩韦肟处理组。将ev-a71 gz-cii按照1.2
×
108pfu的致死剂量腹腔注射感染小鼠,每组10只,取两次的平均值。攻毒6小时前,化合物c、化合物e、化合物k按照0.05mg/kg/d、0.2mg/kg/d剂量口服给药,恩韦肟按照0.2mg/kg剂量口服给药,每天给药2次,连续给药。每天监测小鼠体重变化、发病及死亡情况,持续观察14天,结果见表2。
[0127]
表2、本发明合成化合物的体内抗ev71病毒活性结果
[0128][0129]
试验结果表明,病毒对照组的小鼠全部死亡,化合物c在0.05mg/kg/d、0.2mg/kg/d的给药剂量下小鼠的存活率分别为55%、100%,化合物e在0.05mg/kg/d、0.2mg/kg/d的给药剂量下小鼠的存活率分别为50%、100%,化合物k在0.05mg/kg/d、0.2mg/kg/d的给药剂量下小鼠的存活率分别为60%、100%,效果均优于恩韦肟。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1