一种含有氢键的透明聚酰亚胺及其制备方法和应用
1.本发明属于聚酰亚胺材料制备和应用领域,具体涉及一种高性能透明聚酰亚胺薄膜材料及其制备方法和应用。
背景技术:
2.柔性透明衬底是柔性透明电子器件的重要组成部分,作为柔性透明显示用的衬底需要具备以下几个条件:
3.(1)具有良好的耐热性,无机半导体器件在加工过程中需要较高温度,例如,碳纳米管薄膜晶体管制备需要200℃以上,铟镓锌氧化物(igzo)的加工温度在350℃以上,因此为保证在器件加工过程中柔性基底与其上的电极具有较好的相对尺寸稳定性,基底的热膨胀系数(cte)应该在10ppm/℃以下来使其与其上的电极cte匹配,使其不发生剥离甚至脱落。
4.(2)具有良好的耐有机溶剂性,防止基底在柔性器件加工过程中受有机溶剂腐蚀而发生溶胀甚至溶解,影响器件的尺寸稳定性。
5.(3)具有良好的透光性,500nm以上波长的透光率超过90%。
6.透明聚酰亚胺(pi)由于具有高热稳定性、高光透过率等优点,被认为是柔性显示领域最具潜力的衬底材料。但是,普通pi由于电荷转移显示深黄色,无法满足用于显示器领域的无色透明的性质要求。通过在分子链主链中引入柔性片段降低分子链刚性、将芳香基改为脂肪基降低分子链共轭程度,侧链引入大体积官能团增大分子间距离等方法来破坏分子间电荷转移作用常被用来制备透明聚酰亚胺。但是以上这些方法的引入会使得聚酰亚胺分子链的刚性降低,从而导致玻璃化转变温度(tg)降低,cte增大。在柔性显示器件制备过程中,由于聚酰亚胺的cte明显大于无机材料,形成的薄膜器件在加工或弯曲过程中容易剥离或脱落。目前,在单体结构中引入
‑
cf3是常用的制备透明聚酰亚胺的方法,但是
‑
cf3的引入使得分子间距离明显增大,溶剂分子容易进入链间,使得聚酰亚胺薄膜的耐溶剂性降低,在柔性显示器件制备过程中常用的有机溶剂(如丙酮)中易溶胀甚至溶解,使其难以作为柔性显示用衬底。因此,如何降低薄膜的cte,提高薄膜的耐溶剂性是解决当前柔性衬底材料的关键问题。
7.目前常用的降低无色聚酰亚胺体系cte的方法有以下几种。第一种是通过双向薄膜拉伸工艺来使得分子链定向排列制备超低热膨胀系数的材料,典型的比如upilex s,通过热处理双轴定向拉伸后,其cte最低可以达到12ppm/℃,但是这种方法对设备要求极为苛刻,生产线复杂,并且cte存在明显的各向异性,无法满足作为柔性基底所需要的各向同性要求。第二种是通过改变二酐或二胺的单体结构来调整聚合物的结构或配比来降低热膨胀系数,cn105175723a就是用了这种方法,但是这种改变之后薄膜黄度较大,无法满足透明性要求。第三种是通过向聚酰亚胺体系中加入无机纳米粒子如二氧化硅等降低聚酰亚胺薄膜的热膨胀系数,例如cn104411744a公开的聚酰亚胺薄膜制备方法,但是该方法降低cte效果不明显(cte最低为12.5ppm/℃),并且添加过多无机纳米粒子会导致发生团聚,进而影响薄
膜性能。因此,如何制备具有高热稳定性和高耐溶剂性,同时具有低雾度和高透明性的聚酰亚胺薄膜是目前亟待解决的问题。
8.在分子链中引入氢键是降低体系cte的一种有效方法,利用氢键使分子间相互作用力显著增强,分子间距离减小,分子运动能力减弱,从而可以使得体系具有较低的cte和较好的耐有机溶剂性。例如,目前已经报道的聚酰亚胺
‑
聚酰胺体系通过聚酰胺部分引入氢键来增加分子链刚度,提高氢键数目,从而降低薄膜的cte,提高耐溶剂性。但是目前聚酰亚胺
‑
聚酰胺结构大多数是通过无规共聚方法合成的,无规共聚形成的氢键排列规整性较差,大大降低了氢键的作用,因此薄膜整体性能较差,cte较大,黄度和雾度都较大。如cn108431090a报道了一种无规共聚的聚酰亚胺
‑
聚酰胺薄膜,cte为52ppm/℃,黄度为9.8,雾度为10。cn108368338a也报道了一种无规共聚的聚酰亚胺
‑
聚酰胺薄膜,其cte为11ppm/℃,黄度为3,雾度为1,耐溶剂性较差,在常见有机溶剂如丙酮和甲苯中会发生变形。其它一些专利文献,如cn109476914a、us20200223983a1等也报道了无规共聚的聚酰亚胺
‑
聚酰胺薄膜。此外,随着酰胺占比的增大,体系会倾向于结晶而使得薄膜雾化,不能很好的起到降低cte和提高耐溶剂性的作用。因此,通过合适的聚合方法在聚酰亚胺体系中引入排列规整的氢键(簇状氢键)是充分发挥氢键作用,增强分子间作用力,增加分子链刚性的必要条件。
技术实现要素:
9.针对目前无色聚酰亚胺薄膜cte较高、耐热性较低、耐溶剂型较差的问题,本发明的技术目的在于提供一种具有低的cte、高耐热性、高耐溶剂性的透明聚酰亚胺薄膜及其制备方法。具体通过嵌段共聚的手段,利用簇状氢键调控聚酰亚胺的热稳定性和耐溶剂性。利用该方法可以制备得到低cte、高耐热性及高耐有机溶剂性的透明聚酰亚胺薄膜。基于此薄膜,可以制备柔性器件,如柔性碳纳米管晶体管器件、柔性氧化物半导体器件和柔性传感器等。
10.在本发明的第一方面,提供了一种低cte、高耐热性及高耐有机溶剂性的含有簇状氢键的透明聚酰亚胺聚合物,其具有式i所示的嵌段结构:
[0011][0012]
其中,m、n、p为正整数,代表聚合单元数目,即聚合度,5≤m≤100,5≤n≤100,p为大于等于1的整数,优选为10≤p≤100。
[0013]
r1、r2、r3、r4相同或不同,各自独立地代表取代或未取代的环烷基、芳基、饱和或不饱和的杂环基,或它们的组合形成的骨架结构。
[0014]
所述环烷基优选为c4
‑
c30,更优选为c4
‑
c24的环烷基,最优选c4~c7的环烷基,例如环丁基、环戊基、环己基等。所述芳基优选为c6
‑
c30,更优选为c6
‑
c24的芳基,例如苯基、萘基、联苯基、菲基、蒽基等。所述饱和或不饱和的杂环基优选为c3
‑
c30,更优选为c3
‑
c12的含有一个或多个相同或不同杂原子的杂环基,其中的杂原子选自氧、氮、硫等,例如环氧乙
烷基、呋喃基、噻吩基、吡咯基、吡啶基、嘧啶基、哒嗪基、吡嗪基、吲哚基、苯并呋喃基、苯并硫代苯基等。
[0015]
上述环烷基、芳基、饱和或不饱和的杂环基上可具有一个或多个取代基,所述取代基优选为c1
‑
c6的烷基、c1
‑
c6的卤素取代烷基、
‑
o
‑
、卤素、氰基、羟基、酰基、亚氨基、磺酰基、苯基或这些基团中的多个组合而成的取代基。
[0016]
由r1和r2组成的第一重复单元(聚酰胺嵌段)由于含有
‑
nh
‑
co
‑
结构,因此可以在两段第一重复单元间形成分子间氢键。通过嵌段共聚的方法可以精确控制m和n的相对比例,从而可以控制簇状氢键的占比来精确控制cte的大小。由r3和r4组成的第二重复单元构成聚酰亚胺结构,使整个聚合物具有耐高温特性和无色透明特性。
[0017]
在式i中,r1在各重复单元中可以相同或不同,各自独立地选自包括但不限于以下结构中的一种(其中波浪线表示连接键,下同):
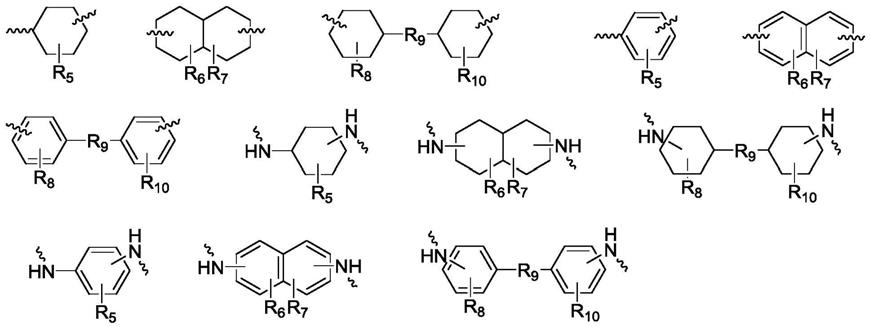
[0018][0019]
以上各结构式中的r9相同或不同,各自独立地选自包括但不限于以下结构中的一种,其中q为正整数,优选为1≤q≤3:
[0020][0021]
以上各结构式中的r5、r6、r7、r8、r
10
相同或不同,各自独立地为h或选自包括但不限于以下结构中的一种:
[0022][0023]
在式i中,r2和r4在各重复单元中可以相同或不同,各自独立地选自包括但不限于以下结构中的一种:
[0024][0025]
其中,上述各结构式中的r
12
相同或不同,各自独立地选自包括但不限于以下结构中的一种(其中,波浪线代表连接键,虚线代表形成环状连接方式,q为正整数,优选为1≤q≤3):
[0026][0027]
其中,r
14
选自包括但不限于以下结构中的一种:
[0028][0029]
上述各结构式中的r
11
、r
13
相同或不同,各自独立地为h或选自包括但不限于以下结构中的一种:
[0030][0031]
为具体说明式i中包含r1和r2的第一重复单元所表示的结构,以二胺单体为4,4
’‑
二氨基
‑
2,2
’‑
双三氟甲基联苯(tfdb)为例进行说明,下面给出几个具体例子。
[0032]
当第一重复单元使用二羰基二氯化合物引入氢键时,可采用二胺与二羰基二氯化
合物缩合聚合实现,其合成原理为一当量二胺的
‑
nh2和一当量二羰基二氯化合物的
‑
cl脱去一当量hcl,形成
‑
nh
‑
co
‑
结构,以下举例说明常见的合成路线和常见结构:
[0033][0034]
当第一重复单元使用含脲基结构的单体引入氢键时,可采用二胺与含脲基结构的单体缩合聚合实现,其合成原理为一当量二胺的
‑
nh2和一当量脲基结构中的=c=o脱去一当量h2o,形成
‑
nh
‑
co
‑
nh
‑
结构,以下举例说明常见的合成路线和常见结构:
[0035][0036]
除了通过引入二羰基二氯化合物或含脲基结构的单体之外,也可以用自身含氢键的二胺单体引入氢键,因此这种二胺结构可以与二羰基二氯化合物或含脲基结构的单体缩合,这样可以在二胺结构内以及二胺和二羰基二氯化合物或含脲基结构的单体缩合处都含有氢键结构,因此可以引入更多的氢键。以下举例说明常见的合成路线和常见结构:
[0037][0038]
此外,也可以用自身含氢键的二胺单体与不含氢键的二酐缩合,这样可以合成自身含氢键的聚酰亚胺部分。
[0039][0040]
在式i中,r3在各重复单元中可以相同或不同,各自独立地选自包括但不限于以下结构中的一种:
[0041][0042]
其中,虚线代表形成环状连接方式;上述不同结构中的r
15
相同或不同,各自独立地选自包括但不限于以下结构中的一种:
[0043][0044]
其中,r
18
选自包括但不限于以下结构中的一种:
[0045][0046]
以上各结构式中的r
16
、r
17
相同或不同,各自独立地为h或选自包括但不限于以下
结构中的一种:
[0047][0048]
式i中包含r3和r4的第二重复单元可以采用一般的聚酰亚胺合成路线,由一当量二胺和一当量二酐基于亲核取代反应原理形成聚酰胺酸,之后经过热亚胺化或化学亚胺化方法脱去一当量水形成第二重复单元聚酰亚胺。以下举例说明常见的合成路线,其中ar和ar1代表不同的芳香基团或者脂肪基团:
[0049][0050]
在式i所示的聚酰亚胺嵌段共聚物中,第一重复单元与第二重复单元的摩尔比即m与n的数值比可以为(0.1~1):1,优选为(0.2~0.4):1。如上所述,引入第一重复单元,可以在共聚物中形成簇状氢键网络的结构,从而降低体系cte和提高耐溶剂性。因此,当第一重复单元的占比太低时,在共聚物中未形成足够多地氢键网络,从而降低体系cte和提高耐溶剂性的效果不显著;而如果第一重复单元的摩尔比太高,则由于聚合物链刚性太强和氢键过多导致薄膜在亚胺化过程中由于聚合物链结晶而雾化,不能得到透明薄膜。
[0051]
此外,利用嵌段共聚的方法可以得到比一般无规共聚的聚酰亚胺更高的分子量。具体地,式i聚酰亚胺聚合物的重均分子量可以为100,000g/mol至1,000,000g/mol,优选为300,000g/mol至500,000g/mol。
[0052]
本发明具有低cte、高耐热性及高耐腐蚀性的含簇状氢键的透明聚酰亚胺嵌段共聚物薄膜,可以通过以下步骤制备:
[0053]
(a)在极性非质子溶剂中加入二胺单体与二酐单体,在0℃
‑
100℃惰性气氛下反应5小时及以上,生成第二重复单元的前驱体聚酰胺酸,其中二胺单体与二酐单体的摩尔比为1:(0.7~1.3),优选为1:(0.9~1.1);所述极性非质子溶剂可以选自以下化合物中的至少一种:n
‑
甲基
‑2‑
吡咯烷酮、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、γ
‑
丁内酯、丙二醇单甲醚、环戊酮、环己酮、醋酸乙酯、甲苯、甲乙酮等。
[0054]
(b)在包含第二重复单元的溶液中加入包含r2结构的二胺单体。
[0055]
(c)当步骤(b)加入的二胺单体溶解完全后,在上述溶液中缓慢加入至少一种引入
氢键的聚合物单体,反应1小时以上,生成第一重复单元,同时获得聚酰胺酸
‑
聚酰胺聚合物。
[0056]
(d)当采用二羰基二氯化合物引入氢键时,由于cl原子会与氢原子生成盐酸,对体系产生影响,因此需要用试剂消除盐酸,反应2小时及以上;所用试剂可选为环氧丙烷、吡啶等。当采用含有脲基基团的大单体来引入氢键时,不会产生盐酸,因此无需消除盐酸这一步骤。
[0057]
(e)采用热亚胺化或化学亚胺化方法将聚酰胺酸
‑
聚酰胺亚胺化为聚酰亚胺
‑
聚酰胺。
[0058]
经过前述(a)至(d)步骤得到聚酰胺酸
‑
聚酰胺溶液,其质量百分比为10%
‑
30%(更优选为13%
‑
30%)、粘度为2000cp
‑
50000cp(更优选为3000cp
‑
30000cp),将其进行热亚胺化或化学亚胺化。其中,热亚胺化通过将聚酰胺酸
‑
聚酰胺浆料涂布在玻璃或者不锈钢基底上,通过程序升温方式来脱去水分子完成亚胺化过程;化学亚胺化方法可以向反应液中加入碱性催化剂和酸酐脱水剂,发生亚胺化反应,通过沉降、过滤、干燥得到聚酰亚胺粉末;随后将其溶解至极性非质子溶剂中,得到聚酰胺酸
‑
聚酰胺浆料,所述的碱性催化剂可以选自吡啶、三乙胺、二乙胺、三甲胺、三丁胺、三辛胺等。所述的酸酐脱水剂可以选自乙酸酐、丙酸酐、马来酸酐、环丁二酐等。
[0059]
在制备薄膜材料时,将聚酰胺酸
‑
聚酰胺溶液浇铸成薄膜,包括但不限于旋涂、刮涂等方式;然后采用梯度升温方法固化薄膜,同时发生热亚胺化,冷却后得到聚酰亚胺
‑
聚酰胺薄膜。优选为以1~10℃/分钟的升温速率在60℃至400℃的范围加热2~10小时,最高温度至少为薄膜tg的90%。
[0060]
在本发明中,在不会负面影响期望效果的情况下,可添加硅烷偶联剂、可交联化合物、无机填料等提高薄膜形貌。如果需要,可以对聚酰胺酸
‑
聚酰胺薄膜进行双向拉伸等工业处理方法进一步提高薄膜性能。
[0061]
本发明提供的技术方案所取得的技术效果至少包含以下几方面:
[0062]
(1)通过控制第二重复单元的投料比和反应时间,可以得到分子量较大的聚酰亚胺片段,从而有效的阻碍第一重复单元由于含有簇状氢键引起的聚合物链结晶,实现薄膜的高光透过率。通过控制第一重复单元与第二重复单元的摩尔比,可以精确控制簇状氢键的数目,进而实现cte的精确调控。
[0063]
(2)由于引入大量簇状氢键,分子刚性大大增加,运动能力减小,因此,由本发明方法制备的嵌段聚酰亚胺薄膜具有更优的热稳定性,具体表现为更低的热膨胀系数(cte<8ppm/℃)和更高的玻璃化转变温度(tg>315℃),因而薄膜在高温下具有更好的尺寸稳定性。低的cte有利于提高薄膜与器件加工过程中使用的无机材料的粘附性,使其不易卷曲或剥离;高的tg保证薄膜在器件加工所需要的高温气氛中不发生尺寸变化甚至熔化。
[0064]
(3)由于引入大量簇状氢键,形成规则排列的氢键网络,分子链运动能力大大减弱,分子间距离大大减小,溶剂难以进入分子链间。因此,由本发明方法制备的聚酰亚胺薄膜具有更优的耐溶剂性能,薄膜在柔性器件加工过程中需要使用的有机溶剂(比如丙酮)中不发生溶胀溶解,耐溶剂指数在0.5%以下,能够作为柔性电子器件衬底使用。
[0065]
(4)基于本发明制备的无色聚酰亚胺薄膜可以适用于柔性电子器件的基底和盖板使用。例如,可以应用于柔性器件的基底,如柔性碳纳米管晶体管器件、柔性氧化物半导体
器件和柔性传感器;也可以作为柔性器件的盖板,如柔性有机发光二极管(oled)屏、柔性太阳能电池等器件。
附图说明
[0066]
图1为本发明提出的含有氢键的聚酰亚胺的化学结构。
[0067]
图2为部分实施例和参考例制备的薄膜的红外光谱比较图。
[0068]
图3为实施例1、参考例1和参考例5制备的薄膜的红外光谱比较图。
[0069]
图4为实施例1、参考例1、实施例3和参考例2制备的薄膜的红外光谱比较图。
具体实施方式
[0070]
以下以形成含有簇状氢键的聚酰亚胺
‑
聚酰胺嵌段共聚薄膜为例,对本发明进行进一步的详细描述,所举实例只用于解释本发明,但不以任何方式限制本发明的范围。
[0071]
实施例1
[0072]
氮气保护下,在配有机械搅拌的三颈瓶中加入4,4
’‑
二氨基
‑
2,2
’‑
双三氟甲基联苯tfdb(1.6172g,5.05mmol),联苯四甲酸二酐bpda(1.4711g,5mmol),保证tfdb与bpda的摩尔比为1.01:1,加入无水二甲基乙酰胺dmac(20ml),使得溶液固含量为13%,室温反应5小时;之后加入tfdb(0.6244g,1.95mmol)和9.5ml dmac,待tfdb完全溶解后,加入对苯二甲酰氯tpc(0.4060g,2mmol),形成凝胶,再加入环氧丙烷(0.6ml,16.8mmol),室温反应3h,得到固含量为13%的聚酰胺酸
‑
聚酰胺溶液,将其转移至0℃冰箱中保存整晚。随后将其刮涂到玻璃板上,以1~10℃/分钟的升温速率在温度60℃至300℃的范围加热2~10小时,冷却至室温后得具有50μm及100μm的透明聚酰亚胺
‑
聚酰胺薄膜。其化学结构如下所示,其中m:n≈2:5,5≤p≤100:
[0073][0074]
实施例2
[0075]
氮气保护下,在配有机械搅拌的三颈瓶中加入4,4
’‑
二氨基
‑
2,2
’‑
双三氟甲基联苯tfdb(1.7773g,5.55mmol),联苯四甲酸二酐bpda(1.6182g,5.5mmol),保证tfdb与bpda的摩尔比为1.01:1,加入无水二甲基乙酰胺dmac(24ml),使得溶液固含量为13%,室温反应5小时;之后加入tfdb(0.4643g,1.45mmol)和9.5ml dmac,待tfdb完全溶解后,加入对苯二甲酰氯tpc(0.3045g,1.5mmol),形成凝胶,再加入环氧丙烷(0.45ml,12.6mmol),室温反应3h,得到固含量为13%的聚酰胺酸
‑
聚酰胺溶液,将其转移至0℃冰箱中保存整晚。随后将其刮涂到玻璃板上,以1~10℃/分钟的升温速率在温度60℃至300℃的范围加热2~10小时,冷却至室温后得具有50μm及100μm的透明聚酰亚胺
‑
聚酰胺薄膜,其化学结构同实施例1,其中m:n≈1.5:5.5,5≤p≤100。
[0076]
实施例3
[0077]
氮气保护下,在配有机械搅拌的三颈瓶中加入4,4
’‑
二氨基
‑
2,2
’‑
双三氟甲基联苯tfdb(1.9374g,6.05mmol),联苯四甲酸二酐bpda(1.7653g,6mmol),保证tfdb与bpda的摩尔比为1.01:1,加入无水二甲基乙酰胺dmac(26.5ml),使得溶液固含量为13%,室温反应5小时;之后加入tfdb(0.3362g,0.95mmol)和9.5ml dmac,待tfdb完全溶解后,加入对苯二甲酰氯tpc(0.2030g,1mmol),形成凝胶,再加入环氧丙烷(0.3ml,8.4mmol),室温反应3h,得到固含量为13%的聚酰胺酸
‑
聚酰胺溶液,将其转移至0℃冰箱中保存整晚。随后将其刮涂到玻璃板上,以1~10℃/分钟的升温速率在温度60℃至300℃的范围加热2~10小时,冷却至室温后得具有50μm及100μm的透明聚酰亚胺
‑
聚酰胺薄膜,其化学结构同实施例1,其中m:n≈1:6,5≤p≤100。
[0078]
实施例4
[0079]
氮气保护下,在配有机械搅拌的三颈瓶中加入4,4
’‑
二氨基
‑
2,2
’‑
双三氟甲基联苯tfdb(2.0976g,6.55mmol),联苯四甲酸二酐bpda(1.9124g,6.5mmol),保证tfdb与bpda的摩尔比为1.01:1,加入无水二甲基乙酰胺dmac(29ml),使得溶液固含量为13%,室温反应5小时;之后加入tfdb(0.1441g,0.45mmol)和9.5ml dmac,待tfdb完全溶解后,加入对苯二甲酰氯tpc(0.1015g,0.5mmol),形成凝胶,再加入环氧丙烷(0.6ml,16.8mmol),室温反应3h,得到固含量为13%的聚酰胺酸
‑
聚酰胺溶液,将其转移至0℃冰箱中保存整晚。随后将其刮涂到玻璃板上,以1~10℃/分钟的升温速率在温度60℃至300℃的范围加热2~10小时,冷却至室温后得具有50μm及100μm的透明聚酰亚胺
‑
聚酰胺薄膜,其化学结构同实施例1,其中m:n≈0.5:6.5,5≤p≤100。
[0080]
参考例1
[0081]
氮气保护下,在配有机械搅拌的三颈瓶中加入4,4
’‑
二氨基
‑
2,2
’‑
双三氟甲基联苯tfdb(2.2416g,7mmol),联苯四甲酸二酐bpda(1.4711g,5mmol),加入无水二甲基乙酰胺dmac(31ml),室温反应5小时。之后加入对苯二甲酰氯tpc(0.4060g,2mmol),形成凝胶,再加入环氧丙烷(0.6ml,16.8mmol),室温反应3h,得到固含量为13%的聚酰胺酸
‑
聚酰胺溶液,将其转移至0℃冰箱中保存整晚。其成膜工艺与实施例1相同。
[0082]
参考例2
[0083]
参考例2与参考例1不同之处为改变tpc加入的摩尔数为1mmol,相应的调整bpda的摩尔数为6mmol,其它条件与参考例1相同。
[0084]
参考例3
[0085]
氮气保护下,在配有机械搅拌的三颈瓶中加入4,4
’‑
二氨基
‑
2,2
’‑
双三氟甲基联苯tfdb(2.2416g,7mmol),联苯四甲酸二酐bpda(2.0595g,7mmol),加入无水二甲基乙酰胺dmac(31ml),室温反应5小时,得到固含量为13%的聚酰胺酸溶液,将其转移至0℃冰箱中保存整晚,其化学结构如下所示,其中,m为大于等于5的整数。其成膜工艺与实施例1相同。
[0086][0087]
参考例4
[0088]
氮气保护下,在配有机械搅拌的三颈瓶中加入4,4
’‑
二氨基
‑
2,2
’‑
双三氟甲基联
苯tfdb(2.2416g,7mmol),加入无水二甲基乙酰胺dmac(26ml),待其完全溶解后,加入对苯二甲酰氯tpc(1.4210g,7mmol),形成凝胶,再加入环氧丙烷(2.1ml,58.8mmol),室温反应3h,得到固含量为13%的聚酰胺溶液,将其转移至0℃冰箱中保存整晚,其化学结构如下所示,其中,n为大于等于5的整数。其成膜工艺与实施例1相同,由于分子链严重结晶,薄膜雾化严重,外观呈现乳白色。
[0089][0090]
参考例5
[0091]
氮气保护下,在配有机械搅拌的三颈瓶a中加入4,4
’‑
二氨基
‑
2,2
’‑
双三氟甲基联苯tfdb(0.8166g,2.55mmol)和联苯四甲酸二酐bpda(0.7365g,2.5mmol),加入无水二甲基乙酰胺dmac(7ml);在另一个配有机械搅拌的三颈瓶b中,氮气保护下,加入4,4
’‑
二氨基
‑
2,2
’‑
双三氟甲基联苯tfdb(1.4250g,4.45mmol)和联苯四甲酸二酐bpda(0.7356g,2.5mmol),加入无水二甲基乙酰胺dmac(9ml)。a、b两瓶同时分别反应5h,之后将b瓶溶液倒入a瓶溶液,用无水二甲基乙酰胺dmac清洗b瓶溶液后倒入a瓶保证之后加入的dmac的量为13.4ml,搅拌5min,之后加入对苯二甲酰氯tpc(0.4060g,2mmol),再加入环氧丙烷(2.1ml,58.8mmol),室温反应3h,得到固含量为13%的聚酰亚胺
‑
聚酰胺溶液,将其转移至0℃冰箱中保存整晚。
[0092]
参考例6
[0093]
参考例6与实施例3的不同之处在于将浆料刮涂到玻璃板上后,以1~10℃/分钟的升温速率在温度60℃至350℃的范围加热2~10小时。
[0094]
参考例7
[0095]
参考例7与参考例2的不同之处在于将浆料刮涂到玻璃板上后,以1~10℃/分钟的升温速率在温度60℃至350℃的范围加热2~10小时。
[0096]
部分实施例和参考例薄膜的红外光谱特性如图2所示:
[0097]
由参考例4可得,当聚酰亚胺部分占比为零,聚合物分子完全为聚酰胺时,分子在3300
‑
3000cm
‑1处的峰强度远高于嵌段聚酰亚胺
‑
聚酰胺聚合物,而参考例3在3300
‑
3000cm
‑1处的峰强度极低,因此,我们猜测在3349cm
‑1处的峰为簇状氢键的峰。由实施例1
‑
4对比可以看出,随着tpc含量增加,嵌段聚酰亚胺
‑
聚酰胺聚合物中簇状氢键的峰强度增加,这与簇状氢键数目增加相对应。
[0098]
如图3所示,参考例5在3349cm
‑1处的峰位于实施例1和参考例1中间,且参考例5与参考例1很接近,可以说明在后半程反应加入与tpc等当量的tfdb使之形成簇状聚酰胺部分,从而形成簇状氢键是导致聚合物在3349cm
‑1处有明显吸收峰的原因。
[0099]
如图4所示,由实施例1和参考例1以及实施例3和参考例2比较可以看出,嵌段聚酰亚胺
‑
聚酰胺聚合物在3349cm
‑1处峰强度明显高于无规共聚的聚酰亚胺
‑
聚酰胺聚合物,而且峰展宽更小,可以说明我们所得到的嵌段聚合物形成了簇状氢键,氢键排列规则,而无规共聚聚合物未形成排列规则的簇状氢键。
[0100]
对在上述实施例和参考例中制备的薄膜评估表1所列特性,并且结果示于表1中。其cte、tg、光透过率、拉伸强度、杨氏模量均按照本领域常用方法测量,测量结果如表1所
示,其中耐溶剂指数的计算按照如下步骤进行:
[0101]
将薄膜在80℃真空烘箱中干燥1小时,然后在五个随机点处测量膜厚度,用anritsu electronic micrometer测量厚度,并且装置的偏差为
±
0.5%或更小。将薄膜再次浸入含有丙酮的烧杯中30分钟,用水洗涤,在80℃真空烘箱中干燥1小时,然后在五个随机点处测量膜厚度。然后,使用溶剂浸渍之前和之后的膜厚度,计算耐溶剂指数。
[0102]
耐溶剂指数(%)=(t0‑
t
30
)/t0×
100
[0103]
其中,t
30
是在将膜浸入丙酮中30分钟之后的膜厚度,t0是在将膜浸入丙酮之前的膜厚度。
[0104]
表1上述各实施例和参考例薄膜性能测试结果
[0105][0106]
对比以上实施例1
‑
4制备聚酰亚胺
‑
聚酰胺嵌段聚合物的数据可以看出,随tpc含量的增加,tg随tpc含量变化无明显变化趋势,在300
‑
330℃之间波动。cte整体呈线性下降趋势。对比实施例1和参考例3可以发现,添加28.5%(tpc/tpc+bpda)的tpc可以使得cte下降3倍左右,具有明显的降低cte的作用。这是由于tpc含量增加时,分子链整体变得更加刚性,分子间氢键的数目也相应增多,分子链间作用力增强,形成更加致密的氢键网络,使得分子链运动困难,并且氢键在较高温度范围内保持活性,因此薄膜在高温下仍然保持较低的cte。
[0107]
对比实施例1和参考例1可以发现,由实施例1制备的聚酰亚胺
‑
聚酰胺嵌段聚合物比参考例1制备的无规共聚的聚酰亚胺
‑
聚酰胺聚合物cte低3倍左右。这可以归结于氢键的密度不同,由实施例1和参考例1中的化学结构可以看出,实施例1的氢键密度更大,分子间密度更强,因此可以起到很好的降低体系cte的效果。
[0108]
对比实施例3和参考例2可以发现,聚酰亚胺
‑
聚酰胺嵌段聚合物的tg明显高于无规共聚的聚酰亚胺
‑
聚酰胺聚合物。再对比参考例6和参考例7可以发现,当亚胺化温度从
300℃提高到350℃时,聚酰亚胺
‑
聚酰胺嵌段聚合物和无规共聚的聚酰亚胺
‑
聚酰胺聚合物光透过率都会下降,且后者下降幅度明显大于前者。这可以归结于聚酰亚胺部分的耐高温性能较好,聚酰胺部分耐高温性能较差,无规共聚聚酰亚胺聚酰胺共聚物的聚酰亚胺部分的链较短,无法有效的阻碍聚酰胺部分结晶,导致无规共聚聚合物的热稳定性明显低于嵌段聚合物,从而光透过率下降更明显。
[0109]
此外,可以发现随tpc含量增大,耐溶剂指数降低,耐溶剂性增大,这是由于氢键的数目随tpc含量的增大而增大,分子间氢键作用力的增强和分子量刚性的增大使得分子间距离减小,溶剂分子难以进入分子链间从而使得耐溶剂性增强。
[0110]
在机械性能和光学性能方面,不同实施例和参考例在拉伸强度和杨氏模量方面差别不大,但是参考例1
‑
3的光透过率明显大于实施例1
‑
4,这是由于嵌段聚合物中氢键明显增加了分子间作用力,使得分子间距离减小,使得链与链之间的电荷转移作用增强,从而使得光透过率下降。
[0111]
综上所述,我们可以通过控制聚酰亚胺与聚酰胺部分的相对含量来实现降低cte的精确控制和耐溶剂性的相应改善,而不明显影响其它光学性能和机械性能。
[0112]
尽管如上所述详细描述了本发明的具体实施方案,但是对于本领域技术人员来说,显而易见的是,具体描述仅是期望的示例性实施方案并且不应当解释为限制本发明的范围。因此,本发明的实质范围由所附权利要求及其等同物限定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1