一种吲哚咔唑衍生物的合成方法及其长余辉材料的制备方法
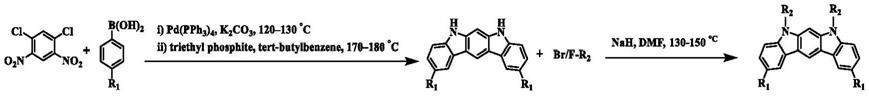
1.本发明涉及有机发光材料技术领域,具体涉及到基于吲哚咔唑和其衍生物的合成及其长余辉材料的制备方法。
背景技术:
2.长余辉,也称之为持续性室温磷光,是指发光材料在撤掉激发源后,依旧可以持续发光几秒钟到几个小时。目前,余辉材料可以大致分为无机材料和有机材料。其中,无机长余辉材料是最早发现,但制造加工方面的困难限制了它们的大规模应用。而有机长余辉材料具有分子修饰方便,可加工性好,成本低,稳定性高和生物相容性好等优点,在加密防伪、生物成像、传感器和有机发光二极管(oled)等领域得到了广泛的应用。
3.对于纯有机分子,受限于较弱的自旋轨道耦合(soc)效应,从第一单重态(s1)到第一三重态(t1)的系统间穿越(isc)效率低,以及包括温度、湿度或分子氧等外部淬灭剂引起的非辐射衰变过程,导致在室温和大气环境下,难以观察到纯有机分子的磷光或余辉,只有在低温和惰性条件才能观察到长余辉现象。通过结晶的方法,从芳基酮、醛、硼酸酯、咔唑等化合物中获得了长余辉。但这不适用于那些难以生长晶体的有机物,以及由于分子间堆积而在固态中具有聚集引起的发射猝灭(acq)问题的分子。另外,受限于形态和后处理,晶体或结晶粉末的长余辉在实际应用过程中存在一定的困难和不便。而通过将有机物掺杂到非晶态聚合物中,组装成非晶态有机余辉材料,可以有效避免上述问题,因此,研发非晶态纯有机余辉材料具有重要意义,并且在实际应用中具有很大的潜力。
4.吲哚咔唑在有机发光材料领域有着广泛应用,到目前为止,关于吲哚咔唑非晶态有机长余辉材料的报道仍然缺乏。另外,吲哚咔唑的研究多集中于两类异构体,其他异构体的合成,应用研究十分稀少,并且相关异构体的合成步骤繁琐、产率低。因此,研发高效、便捷的合成方法是很有必要的,并探索出一种制备吲哚咔唑非晶态纯有机长余辉材料的方法不仅为吲哚咔唑的研究提供新方向,而且扩展其新应用,都具有重要意义。
技术实现要素:
5.本发明的目的在于提供基于吲哚咔唑的合成及其长余辉材料制备方法,可以使吲哚咔唑在室温下实现长余辉。
6.本发明的技术方案是:一种吲哚咔唑衍生物的合成方法,反应式如下:
7.式1:
[0008][0009]
;或式2:
[0010][0011]
;或式3:
[0012][0013]
;或式4:
[0014][0015]
;或式5:
[0016][0017]
;所述的式1
‑
5中r1为h、卤素、为h、卤素、
[0018]
r2为h、c1
‑
c10烷基、
[0019]
进一步的,所述的式3中第一步反应的最佳温度为190℃。
[0020]
进一步的,所述的式5中第一步反应的最佳温度为190℃。
[0021]
一种基于所述的一种吲哚咔唑衍生物的长余辉材料的制备方法,聚乙烯醇水溶液作为主体溶液;吲哚咔唑衍生物溶解于有机溶剂作为客体溶液;将主体溶液加入到客体溶液中,在60
‑
70℃下,超声得到透明混合液,将其滴涂在基板上,然后放入真空干燥箱,得到掺杂吲哚咔唑衍生物的聚乙烯醇薄膜,即长余辉材料。
[0022]
进一步的,吲哚咔唑衍生物的配比浓度在0.1
‑
70wt.%。
[0023]
进一步的,所述的有机溶剂包括甲醇、乙腈、四氢呋喃和二甲基亚砜。
[0024]
进一步的,所述的基板包括石英、玻璃、纸张、塑料、木板、金属和编织物。
[0025]
本发明的有益效果:本发明通过使用新方法合成吲哚咔唑衍生物,大大缩短了反应时间,提高了产率,降低生产成本。与目前有关合成吲哚咔唑的报道相比,本发明合成icz
‑
o所用原料成本较低,以乙酸为反应溶剂,一锅法合成,产率高(专利cn 112250685 a, 2021.01.22.产率89.3%,提高到95.6%);在现有合成icz
‑
m1的技术上优化了合成方案,提高了产率(专利cn 111205313 a,2020.05.29. 第一步产率70%,提高到86.7%),简化合成步骤(文献synth. commun.2000,30,3651
‑
3668.三步减少为两步),缩短反应时间;与已经报道合成icz
‑
m2的技术相比,使用的原料便宜易获取,一锅法(文献j.org.chem.2017,82(24),13583
‑
13593.分三步)合成,反应时间短(由文献synth.commun.2000,30(20),3651
‑
3668.20多小时缩短为5小时);优化了合成icz
‑
p1的技术,减少繁琐的控温步骤,缩短了反应时间(文献new.j.chem.2014,38,2368
‑
2378.需要四步控温环节减少为一步,总反应4
‑
5小时,缩短为2小时);使用新方法合成 icz
‑
p2,以1,4
‑
环己二酮、盐酸苯肼盐为原料,二苯醚作为溶剂一锅法(文献j.org.chem.2017,82,13583
‑
13593.需要三步)合成,所用原料便宜易获取,可直接使用,并且反应时间短、操作简便。
[0026]
并且通过将吲哚咔唑掺杂到聚乙烯醇聚合物中,制备成薄膜,该基质为吲哚咔唑提供了一个刚性环境,可以限制吲哚咔唑分子的运动,从而使吲哚咔唑在室温下实现长余辉发光,该制备方法操作简便,成本低廉;可广泛应用于信息加密、防伪识别、照明等领域。
具体实施方式
[0027]
实施例一
[0028]
本实施例的有机化合物11,12
‑
二氢吲哚并[2,3
‑
a]咔唑(icz
‑
o),化学式为c
18
h
12
n2,分子结构式
[0029]
为:
[0030]
其合成步骤如下:
[0031]
将1,2
‑
环己二酮(1g,8.93mmol)和盐酸苯肼(3.86g,26.78mmol) 依次加入到反应器中,氮气氛围下加入20ml乙酸,搅拌回流24小时后,反应结束,冷却至室温后。将反应混合液缓慢倒入冰水中并搅拌,析出大量黄色固体,将其抽滤并用纯水洗涤,干燥。使用乙酸乙酯/石油醚(1:1)作为洗脱剂通过色谱柱纯化,得到白色固体icz
‑
o (2.18g;95.6%)。其核磁表征数据为1h nmr(400mhz,dmso
‑
d6) δ11.06(s,2h),8.14(d,j=7.7hz,2h),7.90(s,2h),7.68(d,j=8.1hz, 2h),7.42
–
7.36(m,2h),7.21(dd,j=11.1,3.9hz,2h)。
[0032]
实施例二
[0033]
本实施例的有机化合物5,7
‑
二氢
‑
吲哚并[2,3
‑
b]咔唑(icz
‑
m1),化学式为c
18
h
12
n2,分子结构式为:
[0034]
其合成步骤如下:
[0035]
第一步:称取1,5
‑
二氯
‑
2,4
‑
二硝基苯(2.50g,10.55mmol),苯基硼酸(3.21g,31.65mmol),四(三苯基膦)钯(1.83g,1.58mmol) 和k2co3(4.37g,0.368mmol)依次加入到反应器,在氮气氛围下加入乙醇(7ml),甲苯(14ml)和水(7ml)的混合溶剂。搅拌回流24小时后,停止反应,冷却至室温。将反应混合物用水和乙酸乙酯萃取,有机层经无水硫酸镁干燥后再减压下浓缩,使用乙酸乙酯/ 石油醚(1∶3)作为洗脱剂,通过色谱柱法纯化,除去有机溶剂得到黄色粗产物(2.93g,收率86.70%)。
[0036]
第二步:把2.70g粗产物和亚磷酸三乙酯(5.61g,33.75mmol) 叔丁基苯(30ml),在氮气氛围下放入反应器,升温至反应混合液回流,搅拌24小时,反应结束后,冷却至室温。除去有机溶剂,并将粗产物通过色谱柱法提纯,使用乙酸乙酯/石油醚(1:3)作为洗脱剂,除去有机溶剂后得到白色固体icz
‑
m1(1.16g,收率43%);其核磁表征数据为1h nmr(400mhz,dmso
‑
d6)δ11.08(s,2h),8.80(s, 1h),8.14(d,j=7.4hz,2h),7.45
–
7.29(m,5h),7.15(d,j=7.4hz, 2h)。
[0037]
实施例三
[0038]
本实施例的有机化合物5,12
‑
二氢吲哚并[3,2
‑
a]咔唑(icz
‑
m2),化学式为c
18
h
12
n2,分子结构式为:
[0039]
其合成步骤如下:
[0040]
称取1,3
‑
环己二酮(1.50g,13.39mmol)和盐酸苯肼(5.79g, 40.18mmol)放入反应器中,在氮气氛围下加入25ml二苯醚,将反应混合液在190℃下回流5小时。冷却至室温后,除去有机溶剂。粗产物通过色谱柱法纯化,使用乙酸乙酯/石油醚(1∶3)作为洗脱剂,旋转蒸发除去有机溶剂,得到黄色固体icz
‑
m2(1.88g,55%)。其核磁表征数据为1h nmr(400mhz,dmso
‑
d6)δ11.71(s,1h),11.50(s, 1h),8.64(d,j=7.7hz,1h),8.11(t,j=8.9hz,2h),7.62(d,j=8.0hz, 1h),7.56(d,j=8.0hz,1h),7.41(dd,j=11.2,3.9hz,1h),7.35
–
7.26 (m,3h),7.19(t,j=7.2hz,1h);
[0041]
实施例四
[0042]
本实施例的有机化合物5,11
‑
二氢吲哚并[3,2
‑
b]咔唑(icz
‑
p1),化学式为c
18
h
12
n2,分子结构式为:
[0043]
其合成步骤如下:
[0044]
第一步:将1,4
‑
环己二酮(1g,8.93mmol)溶解在10ml乙醇中,加入3滴乙酸,在室温下搅拌20分钟,然后加入盐酸苯肼(3.86 g,26.78mmol),室温下搅拌2小时。然后将反应混合物倒入40ml 纯水中,生产黄色沉淀,过滤并用纯水洗涤,干燥,得深红色固体。
[0045]
第二步:取2.40g中间体,在10℃下加入浓硫酸/乙酸=(4:120 ml)的混合液,加热至30℃保持30分钟,然后升温至60
‑
70℃反应 1小时,最后回流反应2小时,反应结束后,冷却至室温。将反应混合液缓慢倒入50ml水中并搅拌,析出大量黄色固体,将其过滤出并用纯水洗涤,然后真空干燥,使用乙酸乙酯/石油醚(2:1)作为洗脱剂,通过色谱柱提纯,得白色固体产物icz
‑
p1(1.32g,57.90%)。其核磁表征数据为1h nmr(400mhz,dmso
‑
d6)δ11.00(s,2h),8.19 (d,j=7.7hz,2h),8.10(s,2h),7.45(d,j=7.9hz,2h),7.37(t,j=7.6 hz,2h),7.12(t,j=7.4hz,2h)。
[0046]
实施例五
[0047]
本实施例的有机化合物5,8
‑
二氢吲哚[2,3
‑
c]咔唑(icz
‑
p2),化学式为c
18
h
12
n2,分子结构式为:
[0048]
其合成步骤如下:
[0049]
将1,3
‑
环己二酮(1.50g,13.39mmol)和盐酸苯肼(5.79g; 40.18mmol)加入到反应器,氮气氛围下加入25ml二苯醚,然后再 190℃下回流搅拌5小时,反应结束后,冷却至室温。旋转蒸发除去有机溶剂,使用二氯甲烷/石油醚(2:1)作为洗脱剂,通过剂柱色谱法纯化粗产物,得到黄色固体icz
‑
p2(2.02g;59%)。其核磁表征数据为1h nmr(400mhz,dmso
‑
d6)δ11.46(s,2h),8.72(d,j=8.0 hz,2h),7.65(s,2h),7.60(d,j=8.0hz,2h),7.44(t,j=7.6hz,2h), 7.31(t,j=7.5hz,2h)。
[0050]
实施例六
[0051]
本实施例的有机化合物3,8
‑
二溴
‑
11,12
‑
二氢吲哚[2,3
‑
a]咔唑 (m1),化学式为c
18
h
10
br2n2,分子结构式为:
[0052]
其合成步骤如下:
[0053]
将1,2
‑
环己二酮(1g,8.93mmol)和对溴盐酸苯肼(4.99g, 22.32mmol)依次加入到反应器中,氮气氛围下加入20ml乙酸,搅拌回流24小时后,反应结束,冷却至室温后。将反应混合液缓慢倒入冰水中并搅拌,析出大量黄色固体,将其抽滤并用纯水洗涤,干燥。使用乙酸乙酯/石油醚(1:1)作为洗脱剂通过色谱柱纯化,得到白色固体(m1)(3.13g;收率85%)。其核磁表征数据为1h nmr(400 mhz,dmso
‑
d6)δ7.53(d,j=8.7hz,2h),7.71(d,j=8.7hz,2h),7.99 (s,2h),8.43(s,2h),11.33(s,2h).。
[0054]
实施例七
[0055]
本实施例的有机化合物11,12
‑
二丁基
‑
11,12
‑
二氢吲哚[2,3
‑
a]咔唑 (m2),化学式为c
26
h
28
n2,分子结构式为:
[0056]
其合成步骤如下:
[0057]
将11,12
‑
二氢吲哚并[2,3
‑
a]咔唑(icz
‑
o)(1g,3.91mmol)和氢化钠(0.29g,11.72mmol)加入到反应器,量取10ml n,n
‑
二甲基甲酰胺,室温下搅拌反应30分钟,再加入溴代正丁烷(1.61g,11.72 mmol),130
‑
150℃下继续反应3小时,反应结束后,旋转蒸发除去有机溶剂,使用二氯甲烷/石油醚(1:1)作为洗脱剂通过色谱柱纯化,得到黄色固体m2(1.36g;收率95%)。其核磁表征数据为1h nmr (400mhz,cdcl3)δ8.12(d,j=7.7hz,2h),7.94(s,2h),7.55(d,j=8.2 hz,2h),7.46(t,j=7.6hz,2h),7.29(t,j=7.4hz,2h),4.64
‑
4.51(m,4h), 1.70
‑
1.60(m,4h),1.11(dd,j=7.5hz,4h),0.78(t,j=7.4hz,6h)。
[0058]
实施例八
[0059]
称取100mg聚乙烯醇固体,90℃搅拌溶解于1ml纯水中,作为主体溶液。称取1mg5,8
‑
二氢吲哚[2,3
‑
c]咔唑(icz
‑
p2),溶解在 1ml纯甲醇溶剂(或者其他能与水互溶并且能溶解吲哚咔唑的有机溶剂,如乙腈、四氢呋喃,二甲基亚砜等),作为客体溶液。将聚乙烯醇溶液加入到icz
‑
p2溶液中,在65℃下,超声15分钟,得到透明混合液,将其滴涂在石英基板上(或者玻璃、纸张、塑料、木板、金属、编织物等其他载体),然后放入真空干燥箱,在65℃下干燥30 分钟,得到掺杂浓度1wt.%的5,8
‑
二氢吲哚[2,3
‑
c]咔唑(icz
‑
p2)的聚乙烯醇薄膜。用波长365nm紫外灯照射3s,即可用肉眼观察到长达四十多秒的绿色余辉。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1