具有磺酰基联苯结构的金属有机骨架材料及其制备方法和应用以及羧酸酯化的方法与流程
1.本发明涉及金属有机骨架材料技术领域,具体地,涉及一种具有磺酰基联苯结构的金属有机骨架材料及其制备方法和应用以及羧酸酯化的方法。
背景技术:
2.金属有机骨架化合物(metal-organic frameworks),也称配位聚合物,是以无机金属为中心与具有桥联作用的有机配体通过相互连接形成一种具有周期性网络结构的晶态多孔金属有机材料。它不同于传统的无机多孔化合物和有机化合物,其同时具有无机和有机化合物材料的特点,由于其具有高孔隙率、高比表面积,孔道可调控节性、结构多样性等优点,使其在气体储存(储氢)、吸附与分离、多项催化、光电磁等领域具有广泛的应用前景。mofs材料的研究及相关应用已成为国际前沿的研究热点。例如yaghi o.m.et al.science,2013,341,974。
3.酸性金属有机骨架化合物材料的合成,目前主要采用的是以磺化方法为基础的混合搅拌合成法。由于氯磺酸为亲水性磺化物,得到的磺化金属有机骨架化合物材料在反应中很容易吸附水,导致活性和循环性能都不高。因此为了解决以上问题及解决反应要求的多样性,设计合成一种具有强酸性金属有机骨架化合物是具有重要意义的。green chem.,2014,16,2490
–
2499和cn110903489a都是使用mofs材料与磺酸反应生成酸性mofs材料。其对mofs的具体结构无法求证,也无法保证酸密度最大值且不是一个非常固定数值。
4.作为一类重要的有机化工产品和有机化工原料,乙酸酯的工业生产水平及生产能力对我国的化工行业发展具有重要影响。常见的乙酸酯包括乙酸甲酯、乙酸正丙酯、乙酸正丁酯、乙酸异丁酯、乙酸异戊酯等。乙酸酯常用作有机溶剂和香料,也可用于表面活性剂、增塑剂、涂料等。随着人们的环保意识的增强,乙酸酯逐渐成为苯、甲苯、甲己酮等有机溶剂的替代品。工业上生产乙酸酯的传统工艺是以浓硫酸作催化剂,催化乙酸和醇类发生酯化反应生成乙酸酯。浓硫酸催化剂具有价格低的优点,但使用浓硫酸作催化剂环境污染严重、对设备材质要求高、发生的副反应多、副产物多、获得的产物分离提纯比较困难。因此,无机酸催化剂用于酯化反应逐渐被淘汰。近年来,我国乙酸酯生产工艺不断发展,乙酸酯的生产能力不断提高,以固体酸或阳离子交换树脂作为乙酸酯合成反应催化剂得到了很大的发展,在工业化生产上也得到了广泛的应用。固体催化剂在酯化反应中表现出稳定性好、选择性高、成本较低、易于分离等优点。但是,这类催化剂反应速度较慢,酯收率偏低。随着乙酸酯的需求日益增强,绿色环保工艺合成乙酸酯前景广阔。因此,开发性能优异的乙酸酯合成反应催化剂,提高反应效率、抑制副产物生成是未来的重要工作方向。
技术实现要素:
5.本发明的目的是为了克服现有技术中用于合成乙酸酯的催化剂副反应过多、环境污染严重以及固体酸催化剂和酸性阳离子交换树脂催化剂催化活性较差、酯选择性不高等
技术问题。
6.为了实现上述目的,本发明第一方面提供一种具有磺酰基联苯结构的金属有机骨架材料,该金属有机骨架材料的配体由结构如式(1)所示的物质提供,配位金属m选自zr、cr和al中的一种;式(1)中,m’选自h、li、na、k、cs、ca和mg中的一种;
[0007][0008]
本发明第二方面提供一种制备具有磺酰基联苯结构的金属有机骨架材料的方法,该方法包括以下步骤:
[0009]
(1)在第一溶剂和任选的酸的存在下,使式(3)所示的化合物与待配位金属源进行配位反应;
[0010]
(2)任选地,将配位反应的产物进行酸化;
[0011]
其中,式(3)中,m”选自li、na、k、cs、ca和mg中的一种;待配位金属源中的金属选自zr、cr和al中的一种;
[0012][0013]
本发明第三方面提供一种前述第一方面所述的金属有机骨架材料或第二方面所述的方法在酯化反应中的应用,特别是在羧酸酯化中的应用。
[0014]
本发明第四方面提供一种羧酸酯化的方法,该方法包括:在催化剂的存在下,使羧酸与醇接触;所述催化剂为前述第一方面所述的金属有机骨架材料;
[0015]
或者,按照前述第二方面所述方法制备金属有机骨架材料,并在该金属有机骨架材料的存在下,使羧酸与醇接触。
[0016]
与现有技术相比,本发明提供的技术方案至少具有以下优势:
[0017]
(1)本发明提供的金属有机骨架材料具有优异的比表面积、孔容、孔径和酸密度,在用于酯化反应,特别是羧酸与醇的酯化反应时,即使在相对较低反应温度,还能够有效抑制副反应的发生,有效提高羧酸酯化反应的酯化率。
[0018]
(2)本发明还提供了一种制备具有磺酰基联苯结构的金属有机骨架材料的方法,该方法采用先构建有机配体骨架,再与金属中心进行配位的方式,能够使得制备的金属有机骨架材料结构稳定(与传统的酸性树脂催化剂相比热稳定性强,本发明提提供的金属有
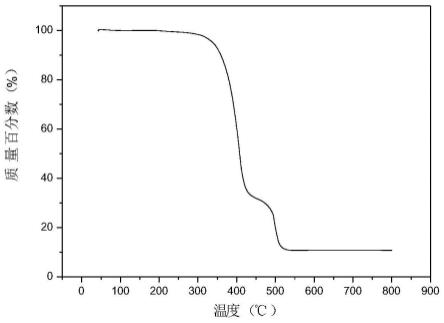
机骨架材料在350℃以下结构稳定,参见图1);并且,本发明提供的方法,反应过程中不存在大量的废酸以及so2等污染物,环境友好。另外,本发明优选的实施方式中,例如制备式(3)所示的化合物时,采用固相的起催化作用的金属卤化物,原料和产物都可以溶解在溶剂中,因此还存在易分离的优势。同时,本发明提供的方法具有步骤简单、操作方便以及可以采用固定床非均相催化的反应器的优势。
附图说明
[0019]
图1是实施例1制备的金属有机骨架材料的热稳定性曲线。
具体实施方式
[0020]
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
[0021]
本发明第一方面提供一种具有磺酰基联苯结构的金属有机骨架材料,该金属有机骨架材料的配体由结构如式(1)所示的物质提供,配位金属m选自zr、cr和al中的一种;式(1)中,m’选自h、li、na、k、cs、ca和mg中的一种;
[0022][0023]
根据本发明的一些实施方式,所述金属有机骨架材料具有如式(2)所示的结构:
[0024][0025]
根据本发明的一些实施方式,所述金属有机骨架材料的比表面积为10-500m2/g,总孔容为1
×
10-3-20
×
10-3
cm3/g,优选为5
×
10-3-20
×
10-3
cm3/g,平均孔半径≤10nm,优选平均孔半径≤2.5nm。
[0026]
优选地,所述金属有机骨架材料的比表面积为100-300m2/g,总孔容为6
×
10-3-10
×
10-3
cm3/g,平均孔半径为0.1-2nm,优选为1.5-2nm。
[0027]
本发明第二方面提供一种制备前述具有磺酰基联苯结构的金属有机骨架材料的方法,该方法包括以下步骤:
[0028]
(1)在第一溶剂和任选的酸的存在下,使式(3)所示的化合物与待配位金属源进行配位反应;
[0029]
(2)任选地,将配位反应的产物进行酸化;
[0030]
其中,式(ii)中,m”选自li、na、k、cs、ca和mg中的一种;待配位金属源中的金属选自zr、cr和al中的一种;
[0031][0032]
根据本发明的一些实施方式,步骤(1)中,所述配位反应的条件可以包括:温度为80-200℃,优选为100-150℃;时间为5-30h,优选为10-24h。
[0033]
根据本发明的一些实施方式,式(3)所示的化合物、待配位金属源、酸以及第一溶剂的摩尔比可以为1:(1.5-5):(0-20):(1-100);优选为1:(2-3):(0-10):(2-50)。
[0034]
本发明中,在配位反应中,加入酸(如乙酸)能够起到模版剂作用,在晶体表面形成催化活性空位,进一步用于后期修饰。
[0035]
根据本发明的一些实施方式,所述待配位金属源可以选自zrcl4、cr(no3)
3 6h2o、alo4(oh)2和zn(no3)2中的一种;优选选自zrcl4、cr(no3)36h2o和alo4(oh)2中的一种。
[0036]
根据本发明的一些实施方式,所述第一溶剂可以选自n,n-二甲基甲酰胺、二甲基亚砜和n-甲基吡咯烷酮中的至少一种,优选为n,n-二甲基甲酰胺。
[0037]
根据本发明的一些实施方式,所述酸可以选自盐酸和/或c1-c4的一元有机酸,优选选自盐酸、甲酸和乙酸中的至少一种。
[0038]
本发明中,对步骤(1)中配位反应的后处理操作没有特别的限定,只要能够满足本发明的需求即可。例如可以按照以下方式进行:将配位反应的物料进行离心抽滤(1000-10000rpm),并用洗涤溶剂(例如依次采用dmf、甲醇)进行洗涤(可以重复2-5次);之后在90-180℃下干燥5-10h即可。其中对洗涤溶剂的种类和用量没有特别的限定,只要能够满足本发明的需求即可。
[0039]
根据本发明的一些实施方式,步骤(1)中,式(3)所示的化合物的制备步骤包括:在第二溶剂和金属卤化物的存在下,使4,4
’‑
联苯二甲酸与氯磺酸盐进行取代反应,得到式(3)所示的化合物。
[0040]
根据本发明的一些实施方式,所述取代反应的条件可以包括:温度为30-100℃,优选为40-80℃;时间为5-30h,优选为10-24h。
[0041]
根据本发明的一些实施方式,4,4'-联苯二甲酸、氯磺酸盐以及金属卤化物的摩尔比可以为1:(0.5-2):(0.01-1),优选为1:(0.8-1.2):(0.05-0.5)。
[0042]
根据本发明的一些实施方式,所述金属卤化物选自rucl3、fecl3、alcl3和pdcl2中的至少一种,优选选自rucl3、fecl3和alcl3中的至少一种。
[0043]
根据本发明的一些实施方式,所述第二溶剂选自二氯甲烷、1,2-二氯甲烷和氯仿中的至少一种;优选为二氯甲烷。
[0044]
根据本发明的一些实施方式,相对于1g的4,4
’‑
联苯二甲酸,所述第二溶剂的用量为20-80ml,优选为30-60ml。
[0045]
其中,对上述取代反应的后处理操作没有特别的限定,只要能够满足本发明的需求即可。优选地,在获得式(3)所示的化合物之前,可以进行如下后处理操作:将取代反应的物料进行抽滤,并用洗涤溶剂(例如依次采用二氯甲烷、甲醇)进行洗涤(可以重复2-5次);之后在60-100℃下干燥5-10h即可。其中,对洗涤溶剂的种类和用量没有特别的限定,只要能够满足本发明的需求即可。
[0046]
根据本发明的一些实施方式,步骤(2)中,所述酸化在无机酸的存在下进行,所述无机酸的浓度为0.1-18.4mol/l,优选1-10mol/l。
[0047]
本发明中,所述无机酸可以选自盐酸、硫酸和硝酸中的至少一种,优选为硫酸。其中,硝酸优选为稀硝酸(浓度可以为0.1-1mol/l)。硫酸优选为稀硫酸(浓度可以为0.1-5mol/l)。
[0048]
根据本发明的一些实施方式,所述酸化的条件可以包括:温度为0℃至100℃,优选为20℃至50℃;时间为5-30h,优选为10-24h。
[0049]
根据本发明的一些实施方式,相对于每摩尔的式(3)所示的化合物,所述无机酸的用量为0.2-10mol,优选为0.4-5mol。
[0050]
本领域技术人员能够理解的是:经酸化后,式(3)中的m”由金属转化为h,从而得到强酸性金属有机骨架材料。
[0051]
本发明中,在获得所述金属有机骨架材料之前,可以进行如下后处理操作:将酸化后的物料体系进行抽滤,得到的固体用水进行洗涤(可以重复2-5次)。其中,对洗涤用的水的用量没有特别的限定。
[0052]
本发明第三方面提供一种前述第一方面所述的金属有机骨架材料或第二方面所述的方法在酯化反应中的应用,特别是在羧酸酯化中的应用。
[0053]
本发明第四方面提供一种羧酸酯化的方法,该方法包括:在催化剂的存在下,使羧酸与醇接触;所述催化剂为前述第一方面所述的金属有机骨架材料;
[0054]
或者,按照前述第二方面所述的方法制备金属有机骨架材料,并在该金属有机骨架材料的存在下,使羧酸与醇接触。
[0055]
根据本发明的一些实施方式,所述接触的条件可以包括:温度为20-200℃,优选为50-150℃;压力为0.1-1mpa,优选为0.2-0.5mpa;时间为20-80h,优选为30-60h;羧酸的重量空速为0.5-10h-1
,优选为1-5h-1
。
[0056]
根据本发明的一些实施方式,所述羧酸和醇的摩尔比可以为1:(1-100),优选为1:(2-10)。
[0057]
所述羧酸选自c2-c20的一元或多元羧酸,优选为c2-c10的一元或多元羧酸。
[0058]
所述醇选自c1-c10的一元醇,优选为c1-c6的一元醇。
[0059]
以下将通过实施例对本发明进行详细描述。
[0060]
以下实施例和对比例用到的原料和溶剂均通过商购获得;
[0061]
比表面积、总孔容和平均孔半径通过氮气物理吸附仪(购自安东帕-康塔仪器,型号autosorb-iq)测试得到。
[0062]
热重数据通过热分析仪(购自mettle-toledo,dga-dsc1热分析仪)测得。
[0063]
实施例1
[0064]
(1)向反应瓶中加入1.04g的rucl3、24.20g的4,4
’‑
联苯二甲酸、16.5氯磺酸钠和1000ml的二氯甲烷,50℃下进行取代反应24h,抽滤,固体用100ml的二氯甲烷洗涤三次,100ml的甲醇洗涤三次,80℃下干燥8h,得到白色粉末(核磁检测确认为式(3)所示的化合物,其中,m”为na)31.6g。之后,将zrcl4、式(3)所示的化合物、dmf以及乙酸按照摩尔比为2.5:1:40:6的比例在120℃下进行配位反应(烧结晶),时间24h,4000rpm下离心抽滤,固体用50ml的dmf洗三次,50ml的甲醇洗三次,固体在105℃下干燥8h,得到的白色固体(金属有机骨架材料)。
[0065]
(2)向步骤(1)得到的白色固体中加入5mol/l的硫酸24ml,在25℃搅拌下进行酸化12h;抽滤,固体用50ml水冲洗三遍得到酸化的金属有机骨架材料(zr为金属中心)。
[0066]
比表面积为246.7m2/g,总孔容=8.154
×
10-3
cm3/g,平均孔半径为1.9nm。
[0067]
实施例2
[0068]
(1)向反应瓶中加入1.04g的rucl3、24.20g的4,4
’‑
联苯二甲酸、16.5g氯磺酸钠和1000ml的二氯甲烷,50℃下进行取代反应24h,抽滤,固体用100ml的二氯甲烷洗涤三次,100ml的甲醇洗涤三次,80℃下干燥8h,得到白色粉末(核磁检测确认为式(3)所示的化合物,其中,m”为na)31.6g。之后,将cr(no3)
3 6h2o、式(3)所示的化合物、dmf以及乙酸按照摩尔比为2.5:1:40:6的比例在100℃下进行配位反应(烧结晶),时间24h,5000rpm下离心抽滤,固体用50ml的dmf洗三次,50ml的甲醇洗三次,固体在120℃下干燥6h,得到的白色固体(金属有机骨架材料)。
[0069]
(2)向步骤(1)得到的白色固体中加入2mol/l的硫酸55ml,在25℃搅拌下进行酸化20h;抽滤,固体用50ml水冲洗三遍得到酸化的金属有机骨架材料(cr为金属中心)。
[0070]
比表面积为265.9m2/g,总孔容=8.095cm3/g,平均孔半径为1.9nm。
[0071]
实施例3
[0072]
(1)向反应瓶中加入1.04g的rucl3、24.20g的4,4
’‑
联苯二甲酸、16.5g氯磺酸钠和1000ml的二氯甲烷,50℃下进行取代反应24h,抽滤,固体用100ml的二氯甲烷洗涤三次,100ml的甲醇洗涤三次,80℃下干燥8h,得到白色粉末(核磁检测确认为式(3)所示的化合物,其中,m”为na)31.6g。之后,将alo4(oh)2、式(3)所示的化合物、dmf以及乙酸按照摩尔比为3:1:50:10的比例在150℃下进行配位反应(烧结晶),时间24h,6000rpm下离心抽滤,固体用50ml的dmf洗三次,50ml的甲醇洗三次,固体在130℃下干燥5h,得到的白色固体(金属有机骨架材料)。
[0073]
(2)向步骤(1)得到的白色固体中加入11mol/l的硫酸5ml,在50℃搅拌下进行酸化6h;抽滤,固体用50ml水冲洗三遍得到酸化的金属有机骨架材料(al为金属中心)。
[0074]
比表面积为276.4m2/g,总孔容=8.106cm3/g,平均孔半径为1.9nm。
[0075]
对比例1
[0076]
向100ml的三口烧瓶中,加入1g的uio-66(zr)、1g的氯磺酸和60ml的二氯甲烷,磁
力搅拌下加热至30℃,再加入0.5g的alcl3作为烷基化催化剂,30℃下反应1h,过滤并洗涤后,80℃下干燥过夜。得白色固体,即为金属有机骨架材料。
[0077]
测试例1
[0078]
以实施例1制备的金属有机骨架材料(zr为金属中心)作为催化剂,评价其在乙酸与异戊醇的酯化反应中的催化性能。将5.0g的金属有机骨架材料(zr为金属中心)装填到内径为8mm的不锈钢固定床反应器中,反应温度为95℃、利用氮气调节反应压力为0.3mpa、乙酸的重量空速为1h-1
、异戊醇与乙酸的摩尔比为2:1,反应时间为50小时。乙酸转化率为99.4%,乙酸异戊酯的选择性为98.2%。
[0079]
测试例2
[0080]
以实施例2制备的金属有机骨架材料(cr为金属中心)作为催化剂,评价其在乙酸与异戊醇的酯化反应中的催化性能。将5.0g的金属有机骨架材料装填到内径为8mm的不锈钢固定床反应器中,反应温度为120℃、利用氮气调节反应压力为0.3mpa、乙酸的重量空速为3h-1
、异戊醇与乙酸的摩尔比为2:1,反应时间为50小时。乙酸转化率为97.7%,乙酸异戊酯的选择性为99.5%。
[0081]
测试例3
[0082]
以实施例3制备的金属有机骨架材料(al为金属中心)作为催化剂,评价其在乙酸与异戊醇的酯化反应中的催化性能。将5.0g的金属有机骨架材料装填到内径为8mm的不锈钢固定床反应器中,反应温度为110℃、利用氮气调节反应压力为0.3mpa、乙酸的重量空速为4h-1
、异戊醇与乙酸的摩尔比为4:1,反应时间为50小时。乙酸转化率为96.9%,乙酸异戊酯的选择性为95.7%。
[0083]
测试例4
[0084]
按照测试例2的方式进行,不同的是,用等量的对比例1制备的金属有机骨架材料代替实施例2制备的金属有机骨架材料。
[0085]
最终,乙酸转化率为87.9%,乙酸异戊酯的选择性为85.5%。
[0086]
测试例5
[0087]
按照测试例2的方式进行,不同的是,用等量的uio-66(zr)金属有机骨架化合物代替实施例2制备的金属有机骨架材料。
[0088]
最终,乙酸转化率为13.3%,乙酸异戊酯的选择性为61.4%。
[0089]
测试例6
[0090]
按照测试例2的方式进行,不同的是,用等量的uio-66(zr)金属有机骨架化合物代替实施例2制备的金属有机骨架材料。
[0091]
最终,乙酸转化率为81.3%,乙酸异戊酯的选择性为83.3%。
[0092]
由以上结果可知,采用本发明金属有机骨架材料(在相同反应条件下和催化剂用量下),用于羧酸酯化时,具有较高的反应活性和选择性。
[0093]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1