一种一步法高效催化CO2转化碳酸二甲酯催化剂的方法
一种一步法高效催化co2转化碳酸二甲酯催化剂的方法
技术领域
1.本发明涉及一种制备催化剂的方法,特别是涉及一种一步法高效催化co2转化碳酸二甲酯催化剂的方法。
2.属于化学化工领域,碳酸酯合成行业。具体而言,涉及一种催化co2一步高效转化为碳酸二甲酯的新型高效强碱性离子液体的合成方法以及基于此催化剂的两段固定床串联反应工艺。
背景技术:
3.化石能源过渡消耗造成大气中的co2、ch4等温室气体含量超标,导致全球变暖、极端天气、海平面持续上升,人类生存环境不断恶化。从“碳中和”与环境保护考虑,将能源终端产物co2作为无毒、廉价、可再生的碳资源,低能耗转化为有价值的化学品具有重要学术价值和应用前景,是人类可持续发展的最优策略。
4.利用co2和煤化工平台化合物甲醇(meoh)直接或间接合成碳酸二甲酯(dmc)受到广泛地关注。 dmc带有微淡香味,是一种无毒、易生物降解且环境友好的大宗化学品。分子中氧原子数多达53%以上,含有丰富的官能团,可以进行多种化学反应。dmc用途广泛,可作为合成五大工程塑料聚碳酸酯的单体、合成农药和医药中间体、绿色溶剂以及锂离子电池溶剂等。还可以在汽油中添加,提高汽油的辛烷值,增强抗震性,显著降低pm2.5和nox排放。
5.dmc合成方法有直接合成法、尿素醇解法、甲醇直接/间接氧化羰基化法以及酯交换法等。其中co2与meoh直接合成dmc过程简单,本质安全,但受热力学限制(
△
rhθ=
‑
27.9 kj/mol,
△
rgθ= 32.6 kj/mol),放热反应且不可自发进行,同时co2化学惰性及副产物水无法及时移除反应体系造成平衡限制等因素,导致dmc收率较低。受益于合成氨的规模化效应,尿素醇解路线原料廉价、易得且生产成本相对较低。但由于中间体氨基甲酸甲酯易于分解成异氰酸等副产物,堵塞反应设备,导致示范过程中装置连续运行稳定性较差,dmc收率较低。co气相/液相氧化羰基化法原料便宜易得,主要生成dmc和水,副产物少。间接氧化羰基化法是meoh通过中间产物亚硝酸甲酯羰基化合成dmc。原料成本低,工艺简单,dmc收率高;但采用含cl催化剂和nox作为氧化剂循环,产生大量含酸废水,有草酸二甲酯、甲酸甲酯等多种含氧化合物副产品。甲醇直接氧化羰基化过程o2直接参与,副产物多,存在安全隐患。以环氧丙烷(po)或环氧乙烷(eo)原料与co2的环加成得到碳酸丙烯酯(pc)和碳酸乙烯酯(ec)后经meoh的酯交换得到dmc并副产1,2
‑
丙二醇或乙二醇,该过程具有反应条件温和,催化活性好,工艺相对简单,产品收率高和纯度高等优点。相比于其他dmc合成路线,酯交换法是更环保、更高效的合成途径,是国内主要的dmc工业生产方法。
6.wen等首次研究了以khco3为催化剂,高温(140 ℃)和高压(co2压力12 mpa)条件下po、co2以及meoh反应原料一步合成dmc。反应6 h,po转化率达到96.87%,但dmc收率仅为16.84%。chun等以氯化胆碱/mgo为催化剂,120 ℃和2.5 mpa下反应6 h,dmc的收率达到65.4%,重复使用4次后dmc收率降到12.58%。chen等以1
‑
丁基
‑3‑
甲基咪唑四氟硼酸盐与甲醇钠复合催化剂,高温150 ℃实现po的转化率达到95%,dmc收率达到67.5%。但离子液体与
甲醇钠粉末不相容且与反应体系无法分离,难以重复使用。tia等采用四丁基溴化铵和叔胺复合催化剂,在高温150 ℃以及较高co2初始压力15 mpa下反应,po的转化率达到98%,dmc的收率达到84%。综上所述,目前以po为反应原料一步法合成dmc都存在反应温度过高,较高的co2初始压力,采用季铵盐或卤素与强碱盐的双组分以上的复合催化剂,难以重复使用。
技术实现要素:
7.本发明的目的在于提出一种一步法高效催化co2转化碳酸二甲酯催化剂的方法,本发明提供了一种单一组分、新型结构、耐高温、高稳定性的强碱性离子液体催化co2一步高效转化为碳酸二甲酯的方法,并基于开发的均相离子液体催化剂,提出催化剂无需分离的两端固定床一步合成碳酸二甲酯新工艺。
8.本发明的目的是通过以下技术方案实现的:一种一步法高效催化co2转化碳酸二甲酯催化剂的方法,所述方法包括以下步骤:环氧丙烷、co2和甲醇为原料反应,均相离子液体催化剂实现高收率得到所述碳酸二甲酯。
9.催化剂无需分离的两段连锁反应新工艺,第一段固定床反应器,催化剂首先催化环氧丙烷和co2合成碳酸丙烯酯;而后,催化剂不分离与甲醇混合降温后进入第二段反应
‑
精馏塔,塔顶采出碳酸二甲酯
‑
甲醇共沸物,塔釜为1,2
‑
丙二醇,实现co2和环氧丙烷一步高收率合成碳酸二甲酯;所述催化剂为离子液体;所述离子液体包括阳离子和阴离子;所述阴离子和阳离子均含有含氮杂环。
10.所述的一种一步法高效催化co2转化碳酸二甲酯催化剂的方法,所述反应温度为100~130 ℃、反应时间为2~9 h、co2初始压力1.8~3.8 mpa、催化剂用量为原料po质量0.5~10%;酯交换过程中反应温度为68 ℃;优选地,所述反应温度为120 ℃;优选地,所述反应时间为9 h;优选地,所述co2初始压力2.6 mpa;优选地,所述催化剂用量为原料po质量3%;优选地,所述酯交换反应温度为68 ℃;优选地,所述阳离子具有式或式所示的结构;所述阴离子具有式、式或式所示的结构;
其中,r1独立地选自c1
‑
c6的烷烃基、c2
‑
c6的烯烃基、c3
‑
c6的芳烃基中的一种;优选地,所述含有环氧丙烷、二氧化碳和甲醇的原料中环氧丙烷、二氧化碳和甲醇摩尔比为1:1:10~1:2.8:15;优选地,所述含有环氧丙烷、二氧化碳的原料中环氧丙烷、二氧化碳的摩尔比为1:1~1:2.8;优选地,所述含有碳酸丙烯酯烯酯和甲醇的原料中碳酸丙烯酯和甲醇的摩尔比为1:10;优选地,所述反应的时间为2小时~9小时;优选地,120℃下,反应9 h以内达到化学平衡;优选地,68℃下,反应30 min达到化学平衡;优选地,r1、r2独立地选自
‑
ch3、
‑
ch2ch3、
‑
(ch2)2ch3、
‑
(ch2)3ch3中的一种。
11.所述的一种一步法高效催化co2转化碳酸二甲酯催化剂的方法,所述催化剂为离子液体。
12.所述的一种一步法高效催化co2转化碳酸二甲酯催化剂的方法,所述离子液体的制备包括以下步骤:a1)向含有离子液体阴离子源的溶液i中,加入碱,反应,得到离子液体阴离子金属盐;a2)将所述离子液体阴离子金属盐溶解在溶剂中,加入离子液体阳离子盐,反应,得到所述离子液体。
13.所述的一种一步法高效催化co2转化碳酸二甲酯催化剂的方法,步骤a1)中,所述溶液i中的溶剂选自乙醇、苯、甲苯、二甲苯中的至少一种;所述碱为有机碱或无机碱;所述有机碱包括甲醇钠、乙醇钠、叔丁醇钠、甲醇钾、乙醇钾或叔丁醇钾;所述无机碱包括氢氧化锂、氢氧化钠、氢氧化钾和氢氧化铯;所述离子液体阴离子金属盐选自离子液体阴离子li盐、阴离子na盐、阴离子k盐、离子液体阴离子cs盐中的至少一种;优选地,步骤a1)中,所述溶液i中,离子液体阴离子源的浓度为0.05~0.8 g/ml;所述离子液体阴离子源与所述碱的摩尔比为0.9~1.1;优选地,步骤a1)中,所述离子液体阴离子源包括咪唑、吡咯或者吗啉;优选地,步骤a1)中,所述反应的条件为:冰浴下反应24小时;优选地,步骤a1)还包括:反应结束后,除去溶剂,得到咪唑阴离子盐、吡咯阴离子盐或者吗啉阴离子盐;优选地,步骤a2)中,所述溶剂包括带水剂;所述带水剂选自甲醇、乙醇、苯、甲苯、二甲苯中的至少一种;
所述离子液体阳离子盐选自1
‑
r1‑3‑
甲基
‑
咪唑溴盐、1
‑
r1‑3‑
甲基
‑
咪唑碘盐、n
‑
甲基
‑
n
‑
r2‑
吗啉溴盐、n
‑
甲基
‑
n
‑
r2‑
吗啉碘盐中的至少一种;优选地,步骤a2)中,所述离子液体阴离子金属盐与溶剂的比例为0.1~0.9:0.05~1.2 g/ml;优选地,步骤a2)中,所述反应的条件为:室温下反应24 h;优选地,步骤a2)还包括:反应结束后,除去溶剂,得到所述高纯度离子液体。
14.所述的一种一步法高效催化co2转化碳酸二甲酯催化剂的方法,所述方法工艺中包括催化剂无需分离的两段连锁反应工艺;第一段固定床反应器,催化剂首先催化环氧丙烷和co2合成碳酸丙烯酯;而后,催化剂不分离与甲醇混合降温后直接进入第二段反应
‑
精馏塔,塔顶采出碳酸二甲酯
‑
甲醇共沸物,塔釜为1,2
‑
丙二醇,实现co2和环氧丙烷一步高收率合成碳酸二甲酯。
15.本发明的优点与积极效果是:1.本发明所提供的一种高效一步催化co2转化为碳酸二甲酯催化剂及工艺方法,合成的新结构离子液体具有耐高温、高稳定性以及强碱性的特点,可以高效催化环加成和酯交换反应。
16.2.本发明所提供的一种催化剂无需分离的两段连锁反应工艺可以显著缩短反应流程和降低生产能耗。
附图说明
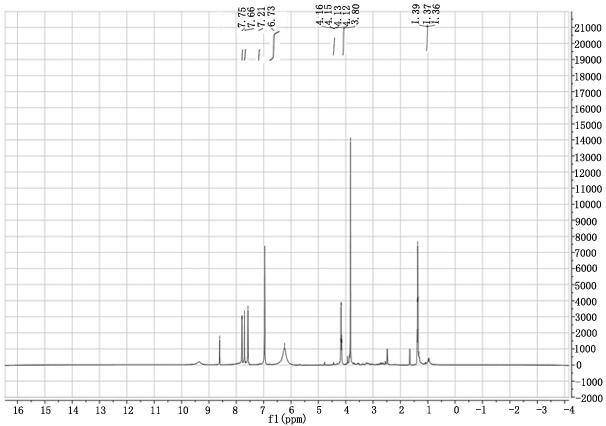
17.图1(a)为本发明实施例1合成的咪唑钾照片;图1(b)为本发明实施例1合成的[emim]im离子液体照片;图2为本发明实施例1合成的[emim]im离子液体核磁h谱;图3为本发明实施例1合成的[emim]im离子液体核磁c谱;图4为本发明实施例3合成的[emim]im离子液体红外图;图5为本发明实施例4合成的[emim]im离子液体热重
‑
差热图;图6为本发明实施例5单组分离子液体催化co2一步合成dmc反应效果;图7为本发明实施例6 ki、tbab以及[emim]im催化po环加成反应效果;图8为本发明实施例6 ki、tbab以及[emim]im催化碳酸酯酯交换反应效果;图9为本发明实施例7反应温度对环加成反应的影响;图10为本发明实施例8反应时间对环加成反应的影响;图11为本发明实施例9 co2初始压力对环加成反应的影响;图12为本发明实施例10催化剂含量对环加成反应的影响;图13为本发明实施例11不同温度下反应时间对dmc收率的影响;图14为本发明实施例12催化剂的循环利用效果;图15为本发明实施例13推测的环加成和酯交换反应机理。
具体实施方式
[0018]
下面结合附图所示实施例详述本发明,但本发明并不局限于这些实施例。
[0019]
本发明第一段固定床反应器合成的[emim]im离子液体首先催化po和co2合成pc。而后,催化剂不分离与meoh混合降温后进入第二段反应
‑
精馏塔,塔顶采出dmc
‑
meoh共沸物,塔釜为1,2
‑
丙二醇,实现co2和po不经分离一步高收率合成dmc及联产产物1,2
‑
丙二醇。
[0020]
第一段固定床反应器筛选出环加成最佳反应条件:催化剂质量为po质量3%,反应温度120 ℃,co2初始压力为2.6 mpa,反应9 h,po转化率高达97.40%,pc选择性大于99%。而后,反应产物进入第二段反应
‑
精馏塔反应加入理论生成pc摩尔量10倍的meoh进行酯交换,在反应温度68 ℃条件下,dmc收率达到83.63%,选择性达到99%,催化剂达到超高催化合成dmc的事实。催化剂重复使用七次未见明显失活。采用制备的新型强碱性离子液体直接代替碘化钾、四丁基溴化胺、甲醇钾、甲醇钠及常规咪唑类离子液体。[emim]im合成工艺简单且环境友好,具有强碱性、高热稳定性以及极佳的催化活性,在碳酸酯合成领域将具有重要的工业应用前景。
[0021]
所述碳酸二甲酯的制备方法,其特征在于,包括以下步骤:环氧丙烷、co2和甲醇为原料反应,均相离子液体催化剂实现高收率得到所述碳酸二甲酯。
[0022]
催化剂无需分离的两段连锁反应新工艺,第一段固定床反应器,催化剂首先催化环氧丙烷和co2合成碳酸丙烯酯。而后,催化剂不分离与甲醇混合降温后进入第二段反应
‑
精馏塔,塔顶采出碳酸二甲酯
‑
甲醇共沸物,塔釜为1,2
‑
丙二醇,实现co2和环氧丙烷一步高收率合成碳酸二甲酯。
[0023]
所述催化剂为离子液体;所述离子液体包括阳离子和阴离子;所述阴离子和阳离子均含有含氮杂环。
[0024]
所述反应温度为100~130 ℃、反应时间为2~9 h、co2初始压力1.8~3.8 mpa、催化剂用量为原料po质量0.5~10%;酯交换过程中反应温度为68 ℃;优选地,所述反应温度为120 ℃;优选地,所述反应时间为9 h;优选地,所述co2初始压力2.6 mpa;优选地,所述催化剂用量为原料po质量3%;优选地,所述酯交换反应温度为68 ℃优选地,所述阳离子具有式或式所示的结构;所述阴离子具有式、式或式所示的结构;
其中,r1独立地选自c1
‑
c6的烷烃基、c2
‑
c6的烯烃基、c3
‑
c6的芳烃基中的一种;优选地,所述含有环氧丙烷、二氧化碳和甲醇的原料中环氧丙烷、二氧化碳和甲醇摩尔比为1:1:10~1:2.8:15;优选地,所述含有环氧丙烷、二氧化碳的原料中环氧丙烷、二氧化碳的摩尔比为1:1~1:2.8;优选地,所述含有碳酸丙烯酯烯酯和甲醇的原料中碳酸丙烯酯和甲醇的摩尔比为1:10;优选地,所述反应的时间为2小时~9小时;优选地,120℃下,反应9 h以内达到化学平衡;优选地,68℃下,反应30 min达到化学平衡;优选地,r1、r2独立地选自
‑
ch3、
‑
ch2ch3、
‑
(ch2)2ch3、
‑
(ch2)3ch3中的一种。
[0025]
所述催化剂为离子液体;所述离子液体的制备方法包括以下步骤:a1)向含有离子液体阴离子源的溶液i中,加入碱,反应,得到离子液体阴离子金属盐;a2)将所述离子液体阴离子金属盐溶解在溶剂中,加入离子液体阳离子盐,反应,得到所述离子液体。
[0026]
所述溶液i中的溶剂选自乙醇、苯、甲苯、二甲苯中的至少一种;所述碱为有机碱或无机碱;所述有机碱包括甲醇钠、乙醇钠、叔丁醇钠、甲醇钾、乙醇钾或叔丁醇钾;所述无机碱包括氢氧化锂、氢氧化钠、氢氧化钾和氢氧化铯;所述离子液体阴离子金属盐选自离子液体阴离子li盐、阴离子na盐、阴离子k盐、离子液体阴离子cs盐中的至少一种;优选地,步骤a1)中,所述溶液i中,离子液体阴离子源的浓度为0.05~0.8 g/ml;所述离子液体阴离子源与所述碱的摩尔比为0.9~1.1;优选地,步骤a1)中,所述离子液体阴离子源包括咪唑、吡咯或者吗啉;优选地,步骤a1)中,所述反应的条件为:冰浴下反应24小时;优选地,步骤a1)还包括:反应结束后,除去溶剂,得到咪唑阴离子盐、吡咯阴离子盐或者吗啉阴离子盐;优选地,步骤a2)中,所述溶剂包括带水剂;所述带水剂选自甲醇、乙醇、苯、甲苯、二甲苯中的至少一种;所述离子液体阳离子盐选自1
‑
r1‑3‑
甲基
‑
咪唑溴盐、1
‑
r1‑3‑
甲基
‑
咪唑碘盐、n
‑
甲基
‑
n
‑
r2‑
吗啉溴盐、n
‑
甲基
‑
n
‑
r2‑
吗啉碘盐中的至少一种;优选地,步骤a2)中,所述离子液体阴离子金属盐与溶剂的比例为0.1~0.9:0.05~
1.2 g/ml;优选地,步骤a2)中,所述反应的条件为:室温下反应24 h;优选地,步骤a2)还包括:反应结束后,除去溶剂,得到所述高纯度离子液体。
[0027]
所述催化剂无需分离的两段连锁反应新工艺,第一段固定床反应器,催化剂首先催化环氧丙烷和co2合成碳酸丙烯酯。而后,催化剂不分离与甲醇混合降温后直接进入第二段反应
‑
精馏塔,塔顶采出碳酸二甲酯
‑
甲醇共沸物,塔釜为1,2
‑
丙二醇,实现co2和环氧丙烷一步高收率合成碳酸二甲酯。
[0028]
作为一种实施方式,所述催化剂为均相强碱离子液体。
[0029]
所述的均相强碱性离子液体催化剂制备方法:(1)咪唑钾盐合成:34 g(0.5 mol)咪唑室温溶解于500 ml无水乙醇中,冰浴条件下将28 g(0.5 mol)koh粉末缓慢加入,并持续搅拌24 h至完全溶解,将上述混合产物过滤,滤液80 ℃减压旋转蒸馏1 h,分离生成的水和乙醇溶剂,降至室温后得到褐色固体咪唑钾盐,经计算咪唑钾收率达到96.62%。
[0030]
(2)1
‑
乙基
‑3‑
甲基
‑
咪唑咪唑盐[emim]im合成:取57.3 g(0.23 mol)含1
‑
乙基
‑3‑
甲基咪唑溴盐质量分数为75%的甲醇溶液,加入200 ml无水乙醇,室温搅拌20 min。缓慢加入23.85 g(0.23 mol)咪唑钾并控制温度为25 ℃附近,生成大量的kbr白色沉淀,持续搅拌24 h后过滤沉淀。压力
‑
0.1mpa温度65 ℃旋转蒸馏2 h,得到黏稠状黄色液体38.92 g,得到1
‑
乙基
‑3‑
甲基
‑
咪唑咪唑盐[emim]im,经计算收率为95.07%。
[0031]
所述的咪唑类离子液体具有如下反应式所述的均相催化剂为高纯度活性离子液体,纯度大于99%。
[0032]
本发明中,“[emim]im”指1
‑
乙基
‑3‑
甲基咪唑咪唑盐。
[0033]
本发明中,“po”指环氧丙烷。
[0034]
本发明中,“meoh”指甲醇。
[0035]
本发明中,“pc”指碳酸丙烯酯。
[0036]
本发明中,“dmc”指碳酸二甲酯。
[0037]
本发明中,“pg”为1,2
‑
丙二醇。
[0038]
本发明中,“pm1”为1
‑
甲氧基
‑2‑
丙醇。
[0039]
本发明中,“pm2”为2
‑
甲氧基
‑1‑
丙醇。
[0040]
本发明中,“hpmc”为1
‑
羟基
‑2‑
丙基甲基碳酸酯。
h后过滤沉淀。压力
‑
0.1mpa温度65 ℃旋转蒸馏2 h,得到如图1 (b) 黏稠状黄色液体38.92 g,经计算收率为95.07%。其核磁谱图如图2和图3所示。图2合成的[emim]im离子液体核磁h谱:1h nmr (500 mhz, dmso
‑
d6) δ 7.75 (s, 1h), 7.66 (s, 1h), 7.21 (s, 1h), 6.73 (s, 3h), 4.14 (q, j = 7.3 hz, 2h), 3.80 (s, 3h), 1.37 (t, j = 7.3 hz, 3h);图3合成的[emim]im离子液体核磁c谱:
13
c nmr (126 mhz, dmso) δ 136.99, 136.18, 123.97, 122.31, 44.51, 36.06, 15。
[0051]
实施例2对1
‑
乙基
‑3‑
甲基
‑
咪唑咪唑离子液体([emim]im)物理参数进行测量,其密度约为1.1 g/ml,粘度为45.6 mpa
· s,利用hammett 指示剂法测定离子液体[emim]im碱强度18.4 < h
‑
< 22.3,该过程中用到的hammett指示剂有:酚酞(9.8)、2,4
‑
二硝基苯胺(15.0)、对硝基苯胺(18.4)、二苯胺(22.3)和苯胺(27.0),根据指示剂颜色变化确定其碱强度区间。结果表明与工业叔丁醇钠碱强度相当,其碱强度高于甲醇钠。根据加入0.1 mol/l苯甲酸的量计算离子液体的碱量为2.83 mmol/g。
[0052]
实施例3合成离子液体1
‑
乙基
‑3‑
甲基咪唑咪唑[emim]im官能团表征结果如图4所示,3142、3074以及2976 cm
‑1处的吸收峰为咪唑环上各取代烷基c
‑
h伸缩振动峰。1668和1570 cm
‑1处的吸收峰归属于咪唑环内的c=n伸缩振动峰,1448 cm
‑1为咪唑环内c=c双键的伸缩振动峰,1172 cm
‑1为咪唑环内的c
‑
n键面内弯曲振动峰,829与763 cm
‑1处的吸收峰分别归属于咪唑环上c
‑
h键的面内和面外弯曲振动。
[0053]
实施例4合成离子液体1
‑
乙基
‑3‑
甲基咪唑咪唑[emim]im氮气氛围下的热稳定性分析如图5所示,有三段失重过程,从室温到144 ℃有2%失重为制备离子液体过程中残留的甲醇溶剂;144至262 ℃出现明显失重,失重量为68.02%,并伴随强吸热,为咪唑环开始大部分的分解;262至356 ℃为咪唑环的完全分解,失重率为26.69%并伴随吸热现象;继续升温制500 ℃,剩余约2%重量保持不变,为[emim]im分解后的积炭。通过实施例1
‑
4所制备的离子液体物理性质、官能团特征以及核磁表征等结果可以断定制备的催化剂为1
‑
乙基
‑
3甲基咪唑咪唑盐,hammett滴定、碱量测定以及热重分析证明其具有高碱强度(18.4 < h
‑
< 22.3)、碱量2.83 mmol/g及高热稳定性。
[0054]
实施例5分别以ki,四丁基溴化铵(tbab)以及1
‑
乙基
‑3‑
甲基咪唑咪唑([emim]im)为催化剂进行合成反应,反应条件如上述催化剂评价方法所示,反应结果如图6。ki和tbab仅具有催化co2发生环加成反应能力,反应2 h时po转化率分别为43.88%和31.53%,反应9 h时po转化率分别为98.79%和98.35%;基本没有催化pc和meoh酯交换以及po和meoh醇解的效果,因此两种催化剂对应的pc选择性均大于99%,dmc选择性和收率均小于1%,ton值近似为零。
[0055]
采用 [emim]im催化剂,反应2和9 h,po转化率为86.90%和98.57%,pc选择性为55.35%和43.66%,dmc选择性为23.63%和36.79%,dmc收率为20.34%和36.27%,对应ton值达到20.77和37.04。与ki和tbab相比,[emim]im展现出良好的dmc收率,但仍不满足预期。其原因是:[emim]im具有强碱性,不但催化co2发生环加成反应,同时催化po和meoh发生醇解反应。产物的gc
‑
ms分析表明,体系中确有醇解产物1
‑
甲氧基
‑2‑
丙醇(pm1)和2
‑
甲氧基
‑1‑
丙
醇(pm2),且反应2和9 h时的pm1选择性已分别达到12.63%和15.59%,pm2选择性达到为8.39%和3.96%。
[0056]
实施例6在无meoh存在的条件下完成po加成环化反应,而后再进行pc和meoh酯交换反应,应可提高dmc选择性和收率。因此,我们设计了催化环加成和酯交换两段连锁反应法一步合成dmc新工艺。两段连锁反应法一步合成dmc的具体流程如下:首先在第一段的固定床反应器内和无meoh添加的条件下进行[emim]im催化po和co2合成pc;而后,向体系加入必要量的meoh混合降温使其进入第二段的反应
‑
精馏塔进行进一步的反应和分离,塔顶溢出dmc
‑
meoh共沸物,塔釜为1,2
‑
丙二醇,从而实现co2和po不经分离的dmc高收率合成。
[0057]
具体验证实验使用ki、tbab和[emim]im催化剂催化,在不添加任何溶剂和助剂的条件下,先催化co2和po环加成在低温催化原位生成的pc与meoh进行酯交换。环氧化反应条件:催化剂质量为po质量3%,反应温度120 ℃,co2初始压力为2.6 mpa。反应2 h及9 h后产物的气相色谱分析结果如图5所示。在反应2 h时三种催化剂的po转化率分别为37.49%、46.37%和64.90%,说明[emim]im催化co2和po环氧化反应能力明显优于ki和tbab。3种催化剂的pc选择性均达99%,说明在该反应条件下,无其他副反应发生。在反应9 h时三种催化剂的po转化率分别为98.28%、97.95%和97.40%,对应的pc选择性仍然维持在99%以上,说明9 h的反应时间足以评价本研究所使用的3种催化剂的最大催化性能。
[0058]
而后,直接加入理论生成pc摩尔量10倍的meoh,反应温度68 ℃,以固定采样间隔在1
‑
120 min时间范围采集反应产物并分析,其结果见图7。显而易见,三种催化剂催化的pc酯交换能力差异明显,tbab活性最差,反应120 min时的pc转化率、dmc选择性和收率仅为33.67%,34.67%和11.68%;ki具有较高的dmc选择性,随着反应时间延长,dmc收率逐渐增加,反应120 min时的pc转化率、dmc选择性和收率仅为26.63%、67.41%和17.96%;[emim]im具有极佳的pc酯交换催化能力,仅反应5 min pc转化率、dmc选择性和收率就高达72.87%、97.36%和70.94%,30 min内达到反应平衡,此时dmc收率为83.63%。增加meoh含量至pc摩尔含量的20倍,反应120 min时的dmc选择性和收率分别为99.84%和87.81%。该过程dmc收率主要受反应平衡限制(原料组成),如果耦合反应
‑
精馏工艺,dmc收率(以co2计算)可以达到98%以上。
[0059]
实施例7反应温度对po与co2环加成反应的影响如图9所示。催化剂[emim]im质量为po质量的3%,初始co2压力为2.6 mpa,以co2/po摩尔比计为1.6:1,反应时间9 h。所有反应温度条件下,目标产物pc选择性均大于99%,说明合成的[emim]im具有优秀的定向催化po环加成能力。100 ℃时,pc收率已达到80.69%,ton值达82.42;持续提高反应温度至110和120 ℃时,pc收率进一步增加至89.36%和97.40%,ton增加至91.28和99.49;继续升高至130 ℃,pc收率为97.75%,ton为99.85,均没有明显变化。考虑到po与co2环加成反应为体积缩小的强放热反应,降低反应温度有利于促进反应向正方向进行,提高po平衡转化率。因此,后续实验优选120 ℃反应条件。
[0060]
实施例8图10展示了反应时间对po与co2环加成反应的影响。催化剂[emim]im质量为po质量的3%,反应温度120 ℃,co2初始压力为2.6 mpa。所有反应时间目标产物pc选择性均大于
99%。当反应时间为2 h,pc收率为64.90%;增加反应时间至4和6 h,pc的收率增加至88.77%和96.20%;增加时间至9 h,pc的收率达到97.40%。由于pc选择性在采样时间区间均在99%以上,所以ton显示了与收率类似的随反应增加趋势,从2 h的63.23增加到9 h的99.49。
[0061]
实施例9co2初始压力对环加成反应的影响如图11所示。催化剂[emim]im含量为po质量的3%,反应温度120 ℃,反应时间9 h。co2不同初始压力下,pc选择性均大于99%。当co2初始压力为1.8 mpa,即co2与po原料摩尔比为1:1时,pc收率为87.61%,ton为89.49;co2初始压力为2.0 mpa(co2/po摩尔比=1.2:1)时,pc的收率为90.94%,ton为92.89;当co2初始压力为2.4 mpa(co2/po摩尔比=1.4:1)时,pc的收率为97.03%,ton为99.11;持续提高co2初始压力至2.6 mpa(co2/po摩尔比=1.6:1)时,pc收率明显增加至97.40%,ton值增加至99.49;继续提升co2压力至3.2(co2/po摩尔比=2.2:1)和3.8 mpa(co2/po摩尔比=2.8:1)时,pc收率反而下降至93.96%和93.25%,ton也降为95.98和95.25。这与很多文献报道的结果一致,在低压过程,pc收率随co2压力升高而增大,co2分压超过一定压力时,pc收率随co2压力升高而缓慢降低。过高的初始压力会导致co2液化,并溶解于反应原料po,稀释催化剂浓度,减弱po与催化剂活性位点之间的作用,降低催化剂的催化效率,不利于反应向正方向进行。因此,后续实验工作优选co2初始压力2.6 mpa。
[0062]
实施例10图12表明了催化剂用量对po与co2环加成反应的影响。co2初始压为2.6 mpa,反应温度120 ℃,反应时间9h。不同催化剂用量,pc选择性均大于99%。当催化剂[emim]im质量为po质量的0.5%时,pc的收率为66.34%,ton值高达406.59;持续增加催化剂用量至po质量的1%和3%时,pc收率明显增加至73.49%和97.40%,而ton值降为225.21和99.49;持续增加催化剂用量至po质量的5%和10%,pc收率达到98.58%和98.95%,提升不明显。即使10%的催化剂含量其ton值仍可以达到30.32。考虑经济性因素,催化剂用量为po质量的3%。
[0063]
实施例11图13考察了[emim]im催化剂在不同反应温度和时间对pc与meoh酯交换反应效率的影响。催化剂用量为反应原料总质量的0.4 wt%,pc/meoh摩尔比为1/10。由于[emim]im催化酯交换反应效率极高, dmc选择性在所有温度条件下均达99%以上,基本没有中间产物生成,所以dmc收率近似于pc选择性。反应温度0、25、50以及68 ℃时反应10 min, dmc收率分别7.95%、10.21%、21.33%、38.01%;反应时间为60 min时,dmc收率分别提高到18.57%、27.27%、58.74%、71.99%。25 ℃时,200 min达到反应平衡;50 ℃时,120 min达到反应平衡;68 ℃反应,60 min内达到平衡。当反应温度从0升高到68 ℃,反应时间30 min时,ton值和dmc的收率得到显著提高,分别从12.96增加到70.15和从12.81%到68.98%。以上实验结果充分说明pc与meoh酯交换反应速率对反应温度有明显的依赖关系,,即使在极端低温条件下,[emim]im也显示了极高的催化活性,能在pc开环后极短的时间内完成酯交换反应,具有超强的酯交换催化能力。
[0064]
实施例12研究 [emim]im离子液体催化剂连续7次高温催化co2和po环氧化反应以及低温催化原位生成pc与meoh的酯交换反应效果。环氧化反应条件:[emim]im含量为po质量3%,环氧化反应温度120 ℃, co2初始压力2.6 mpa(co2/po摩尔比=1.6:1),反应时间9 h,采样分析
时间点9 h。而后,向系统加入理论生成pc摩尔量10倍的meoh,在68 ℃,反应30 min后取样分析。整个反应过程中未添加任何溶剂和助剂,仅用反应初始时加入的同一质量的离子液体[emim]im连续催化环氧化和酯交换两步反应。单次两步反应后,140
‑
145 ℃减压分离meoh、pc、dmc以及1,2
‑
丙二醇等原料和产物,回收剩余的离子液体直接供下一次两段反应循环使用,直到第6个循环结束。
[0065]
po和pc转化率以及dmc收率与离子液体[emim]im重复使用次数的关系如下图14所示。[emim]im首次使用时,po转化率为97.40%、pc转化率85.07%、dmc收率为83.63%。催化剂重复使用一次时, po转化率不仅未降反增加至98.05%,其原因是产物1,2
‑
丙二醇在离子液体中少量残留,通过氢键作用促进po开环。此时pc转化率和dmc收率分别为和84.25%,与首次使用时dmc收率几乎接近,并未出现下降趋势。增加重复使用次数至第四次时,po转化率降为97.34%,dmc收率降为83.82%,但下降趋势并不明显。重复使用5次时,po转化率降为96.68%,dmc收率为80.96%,与首次相比po转化率下降0.72%,dmc收率下降约4%。这里需要强调的是,每次循环取两次产物进行色谱分析,约产生1%离子液体质量损失,依次推算,第5次重复使用时,系统的离子液体实际质量约为第一次的96%。所以,循环使用6次后,额外补充新离子液体0.024 g。于是,第7次反应的po转化率, pc转化率和dmc收率均有回升,分别恢复到了97.48%,84.58%,和83.74%,催化效果与首次使用的新鲜催化剂相当。以上结果证明本研究合成的 [emim]im新结构离子液体具有良好的热稳定性和催化稳定性,重复使用过程中一直能维持较高的环加成和酯交换反应效率,活性没有明显降低。
[0066]
实施例13推测反应机理如图15所示,在反应体系中[emim]im以阴、阳离子的状态存在,咪唑阳离子亲电进攻环氧丙烷的氧原子,使c
‑
o键拉长变形;咪唑阴离子亲核进攻环氧丙烷中空间位阻较小的c原子,使c
‑
o键断裂,形成中间体a。阴阳离子共同作用活化环氧丙烷使其开环,形成含氧负离子的中间物种b。随后中间物种b中氧负离子亲核进攻co2分子中带正电荷的c原子生成碳酸盐中间体c;随后碳酸盐中间体c在咪唑阴离子的作用下进行分子内取代,生成新的c
‑
o键,得到碳酸丙烯酯d,释放1
‑
乙基
‑3‑
甲基咪唑阳离子和咪唑阴离子。
[0067]
酯交换过程中离子液体[emim]im中的咪唑阴离子通过氢键作用捕获甲醇羟基上的氢,进而活化甲醇形成ch3o
−
基团;ch3o
−
亲核进攻pc(d)中的羰基,使c=o键断裂形成中间体e;由于中间体e不稳定,与c相连的四个c—o键至少断裂一条,这是一个典型亲核加成
‑
消去反应。1位c—o键在碱催化体系下无法断裂,是由于c
+
和o
−
不能够以自由基的形式稳定存在;2位c—o键断裂会生成pc和ch3o
−
基团,3和4位c—o键断裂生成中间体f和g。由于f和g结构中与c=o相连o的吸电子作用,导致f和g的电负性弱于ch3o
− 电负性且更容易生成;f和g夺取或者与体系中的h+ 结合生成h中间产物1
‑
羟基
‑2‑
丙基甲基碳酸酯(hpmc)和i中间产物2
‑
羟丙基甲基碳酸酯(hmc);ch3o
−
亲核进攻中间产物hpmc和hmc中的羰基,形成中间体j和k;由于j和k不稳定,4位c—o键断裂形成目标产物dmc和中间产物pg负离子l和m;l和m夺取或者与体系中的h+ 结合生成最终产物pg;pg负离子l和m还可能进攻中间产物hpmc和hmc中的羰基,使c=o键断裂形成中间体n和o;中间体3号位c—o键断裂生成中间产物p和q。
[0068]
实施例14根据实施例13中推测的环加成和酯交换反应机理,合成的新结构离子液体活性中心为强亲和性的咪唑负离子,具有各种支链结构的咪唑、吡咯以及噻吩等阴离子在本专利
保护范围内。
[0069]
以上所述,仅是本发明的几个实施例,并非对本发明做任何形式的限制,虽然本发明以较佳实施例揭示如上,然而并非用以限制本发明,任何熟悉本专业的技术人员,在不脱离本发明技术方案的范围内,利用上述揭示的技术内容做出些许的变动或修饰均等同于等效实施案例,均属于技术方案范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1