一种盐酸帕洛诺司琼水合物晶型及其制备方法与流程
1.本发明涉及盐酸帕洛诺司琼水合物晶型及其制备方法。
技术背景
2.帕洛诺司琼是一种高效、高选择性的5-ht3受体拮抗剂,与传统的5-ht3受体拮抗剂相比,具有疗效高、作用时间长、用量小、不良反应少、耐受性好的优点,主要应用于治疗化疗引起的恶心、呕吐。
3.一些化疗试剂即使使用一次也会导致长达数天的呕吐,需要多次服用止吐药物缓解。目前,帕洛诺司琼、格拉司琼等止吐药通常以静脉注射方式给药,非常不便于患者自行用药,并且注射剂存在着有关活性药物稳定性和储存周期的特殊问题。
4.常见的固体剂型有片剂、胶囊、散剂、滴丸、膜剂等,与液体制剂相比具有物料化学稳定性好,生产制造成本较低,服用与携带方便等特点,在药物制剂中固体制剂约占70%,而原料药的晶型将直接影响固体制剂加工性能,从而影响药物的稳定性、溶解性和生物利用度。因此,药品研发过程中,需要全面研究原料药的晶型。
5.目前,已知盐酸帕洛诺司琼有三种晶型,即晶型ⅰ、晶型ⅱ和无定形晶型,专利wo2008073757a1或us20160214976a1公开了这三种晶型的dsc和粉末衍射表征。其中无定形晶型暴露在空气中极易吸潮,不利于贮存、运输和药物制剂;晶型ⅱ在特定环境中易转化为晶型ⅰ,存在晶型稳定性问题;目前药用晶型主要是晶型ⅰ,且以注射剂为主。
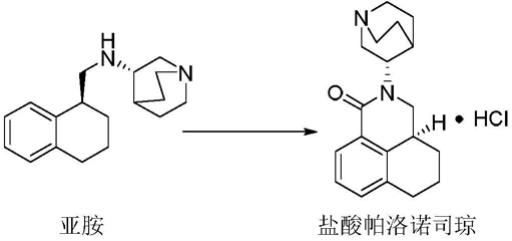
6.盐酸帕洛诺司琼除了上述已知3中晶型外,本技术的发明者在制备盐酸帕洛诺司琼粗品时:
[0007][0008]
发现后处理过程的一种不明有机物析出,对其进行核磁、质谱、高效液相色谱等手段进行鉴别后,确证析出物为盐酸帕洛诺司琼。析出物经有机溶剂重结晶,烘干后得到盐酸帕洛诺司琼水合物晶型。该制备方法简单,成本低廉,易于工业化生产。
技术实现要素:
[0009]
本发明的目的是提供一种盐酸帕洛诺司琼水合物晶型,它具有良好的稳定性,较低的引湿性、更适合用于制备盐酸帕洛诺司琼的固体制剂,对未来的药物的优化和开发具有重要价值。
[0010]
本发明还提供一种盐酸帕洛诺司琼水合物晶型的制备方法。
[0011]
本发明的第一目的:提供一种盐酸帕洛诺司琼水合物晶型,所述水合物晶型用cuka射线测量得到的x射线粉末衍射图中,包括在2θ衍射角为5.6
±
0.2
°
,10.9
±
0.2
°
,13.4
±
0.2
°
,16.9
±
0.2
°
,22.6
±
0.2
°
处的特征衍射峰。
[0012]
进一步地,所述水合物晶型用cuka射线测量得到的x射线粉末衍射图中,还包括在2θ衍射角为12.3
±
0.2
°
,17.9
±
0.2
°
,18.6
±
0.2
°
,19.7
±
0.2
°
,20.8
±
0.2
°
,21.1
±
0.2
°
,22.1
±
0.2
°
,23.0
±
0.2
°
,26.9
±
0.2
°
处特征衍射峰中的至少一个。
[0013]
更具体地,所述盐酸帕洛诺司琼水合物晶型,使用cu-kα辐射,其x射线粉末衍射图谱见附图1,dsc图见附图2,tga图见附图3。
[0014]
从所述水合物晶型的dsc图看见,在66.5℃、227.5℃处有吸热峰。
[0015]
所述水合物晶型的水分含量为5.1%~5.6%。
[0016]
本发明的第二目的:一种盐酸帕洛诺司琼水合物晶型的制备方法,该方法包含如下步骤:
[0017]
a).将(s)-n-(((s)-1,2,3,4-四氢萘-1-基)甲基)奎宁环-3-胺(简称:亚胺),反应溶剂,加入到反应瓶中,加入三光气。加毕,回流反应2小时,加入三氟化硼乙醚。加毕,回流反应6小时。降至室温,加入水,加毕,回流反应4小时,降至0~20℃以下,过滤,使用水洗涤。滤饼,干燥,得盐酸帕洛诺司琼粗品;
[0018]
b).将盐酸帕洛诺司琼粗品使用有机溶剂升温溶解澄清,梯度降温至0~5℃,搅拌2小时,过滤,使用有机溶剂洗涤,干燥,得盐酸帕洛诺司琼水合物晶体。
[0019]
步骤a)中的反应溶剂选自甲苯、二甲苯、氯仿、丙酮、乙酸乙酯、乙酸异丙酯中的至少一种。
[0020]
更进一步地,在上述制备方法中,步骤a中反应溶剂优选甲苯;三光气加入方式优选滴加方式。
[0021]
进一步地,步骤b)中结晶溶剂选自水、甲醇、乙醇、异丙醇、丙酮、乙腈中的一种或几种。
[0022]
步骤b)中的析晶温度为0~30℃。
[0023]
步骤b)中盐酸帕洛诺司琼粗品与有机溶剂的质量比为1:5~35。
[0024]
步骤b)中干燥温度为30~80℃;干燥时间为1~24小时。
[0025]
更进一步地,在上述制备方法中,步骤b中结晶溶剂优选甲醇或乙醇或异丙醇与水的混合体系;结晶溶剂量优选酸帕洛诺司琼粗品与有机溶剂质量比1:5~20;析晶温度优选0~10℃;干燥温度优选40~50℃,干燥5~10小时。
[0026]
本发明由于上述技术方案而产生的积极效果是显而易见的,即本发明提供的盐酸帕洛诺司琼水合物晶型,具有良好的稳定性,较低的引湿性、工艺可开发和易处理等有利性能。本发明提供的盐酸帕洛诺司琼水合物晶型的制备方法简单,成本低廉,易于工业化生产。
附图说明
[0027]
图1为实施例1制备的盐酸帕洛诺司琼水合物晶型的x-射线粉末衍射图谱;
[0028]
图2为实施例1制备的盐酸帕洛诺司琼水合物晶型的dsc图;
[0029]
图3为实施例1制备的盐酸帕洛诺司琼水合物晶型的tga图。
具体实施方式
[0030]
本发明的盐酸帕洛诺司琼水合物晶型的结构表征使用了x射线粉末衍射,差热扫描量热法。化合物纯度使用waters高效液相色谱检测。
[0031]
x射线粉末衍射使用高分辨透射模式xrpd图在panalytical x’pert3x射线粉末衍射分析仪上采集,xrpd测试参数如下:x射线扫描范围(
°
2th)3
°‑
40
°
;扫描步长(
°
2th)0.0131;每步扫描时间(s)97.767。
[0032]
差热扫描量热使用t-a差热热量及dsc2000,操作方法:使用t-zero瓶,加热范围为30~350℃,加热速度为每分钟5℃,氮气流速为80ml/min。
[0033]
下面结合具体的实例对本发明的技术方案作进一步的说明
[0034]
实施例1盐酸帕洛诺司琼水合物晶型型的制备
[0035]
称取(s)-n-(((s)-1,2,3,4-四氢萘-1-基)甲基)奎宁环-3-胺(简称亚胺)20.0g,甲苯200.0g,加入到反应瓶中,室温滴加三光气的甲苯溶液(37.0g三光气加入到150.0g甲苯中搅拌溶解)。滴加完毕,回流反应2小时,加入37.0g三氟化硼乙醚。加毕,回流反应8小时。降至室温,加入水200.0g,加毕,回流反应4~6小时,降至0~20℃,过滤,使用水洗涤,干燥,得盐酸帕洛诺司琼粗品。称取盐酸帕洛诺司琼粗品10.0g,加入无水乙醇140g,水30.0g,加热溶解澄清,降温至10~20℃,搅拌2小时,过滤,使用无水乙醇10.0g洗涤,滤饼于50~60℃,干燥,得盐酸帕洛诺司琼水合物晶型6.25g,收率为62.5%,纯度为99.80%,水含量为5.3%。其xrpd如图1,dsc如图2,tga如图3。
[0036]
实施例2盐酸帕洛诺司琼水合物晶型的制备
[0037]
称取(s)-n-(((s)-1,2,3,4-四氢萘-1-基)甲基)奎宁环-3-胺(简称亚胺)65.0g,二甲苯650.0g,加入到反应瓶中,室温滴加三光气的二甲苯溶液(121.9g三光气加入到487.0g二甲苯中搅拌溶解)。滴加完毕,回流反应2小时,加入122.0g三氟化硼乙醚。加毕,回流反应8小时。降至室温,加入水650.0g,加毕,回流反应4~6小时,降至0~20℃,过滤,使用水洗涤,干燥,得盐酸帕洛诺司琼粗品。称取盐酸帕洛诺司琼粗品50.0g,加入乙醇400.0g,和水200.0g加热溶解澄清,温至0~10℃,搅拌2小时,过滤,使用无水乙醇50.0g洗涤,滤饼于50~60℃,干燥,得盐酸帕洛诺司琼水合物晶型28.1g,收率为56.2%,纯度为99.78%,水含量为5.5%。
[0038]
实施例3盐酸帕洛诺司琼水合物晶型的制备
[0039]
称取(s)-n-(((s)-1,2,3,4-四氢萘-1-基)甲基)奎宁环-3-胺(简称亚胺)40.0g,甲苯400.0g,加入到反应瓶中,分批次加入三光气75.0g。加毕,回流反应2小时,加入75.0g三氟化硼乙醚。加毕,回流反应8小时。降至室温,加入水400.0g,加毕,回流反应4~6小时,降温至0~20℃,过滤,使用水洗涤,干燥,得盐酸帕洛诺司琼粗品。称取盐酸帕洛诺司琼粗品15.0g,加入异丙醇150.0g和水50.0g,加热溶解澄清,梯度降温至0~10℃,搅拌2小时,过滤,使用冷的异丙醇15.0g和水5.0g的混合液洗涤,滤饼于50~60℃,干燥,得盐酸帕洛诺司琼水合物晶型10.05g,收率为67.0%,纯度为99.85%,水含量为5.6%。
[0040]
实施例4盐酸帕洛诺司琼水合物晶型的制备
[0041]
称取(s)-n-(((s)-1,2,3,4-四氢萘-1-基)甲基)奎宁环-3-胺(简称亚胺)20.0g,甲苯200.0g,加入到反应瓶中,室温滴加三光气的甲苯溶液(37.0g三光气加入到150.0g甲苯中搅拌溶解)。滴加完毕,回流反应2小时,加入37.0g三氟化硼乙醚。加毕,回流反应8小
时。降至室温,加入水200.0g,加毕,回流反应4~6小时,降至0~20℃,过滤,使用水洗涤,干燥,得盐酸帕洛诺司琼粗品。称取盐酸帕洛诺司琼粗品10.0g,加入甲醇100g,水20.0g,加热溶解澄清,降温至0~10℃,搅拌2小时,过滤,使用甲醇和水的混合液洗涤,滤饼于40~50℃,干燥,得盐酸帕洛诺司琼水合物晶型4.86g,收率为48.6%,纯度为99.87%,水含量为5.2%。
[0042]
实施例5盐酸帕洛诺司琼水合物晶型的制备
[0043]
称取(s)-n-(((s)-1,2,3,4-四氢萘-1-基)甲基)奎宁环-3-胺(简称亚胺)20.0g,甲苯200.0g,加入到反应瓶中,分批次加入三光气37.5g。加毕,回流反应2小时,加入37.5g三氟化硼乙醚。加毕,回流反应8小时。降至室温,加入水200.0g,加毕,回流反应4~6小时,降温至0~20℃,过滤,使用水洗涤,干燥,得盐酸帕洛诺司琼粗品。称取盐酸帕洛诺司琼粗品10.0g,加入丙酮200.0g,纯净水150.0g,升温溶解,缓慢降温至0~10℃,过滤,使用丙酮和水的混合液洗涤,滤饼于40~50℃,干燥,得盐酸帕洛诺司琼水合物晶型6.8g,收率为68.0%,纯度为99.76%,水含量为5.2%。
[0044]
实施例6盐酸帕洛诺司琼水合物晶型的制备
[0045]
称取(s)-n-(((s)-1,2,3,4-四氢萘-1-基)甲基)奎宁环-3-胺(简称亚胺)20.0g,甲苯200.0g,加入到反应瓶中,分批次加入三光气37.5g。加毕,回流反应2小时,加入37.5g三氟化硼乙醚。加毕,回流反应8小时。降至室温,加入水200.0g,加毕,回流反应4~6小时,降温至0~20℃,过滤,使用水洗涤,干燥,得盐酸帕洛诺司琼粗品。称取盐酸帕洛诺司琼粗品10.0g,加入乙腈150.0g,纯净水200.0g,升温溶解,缓慢降温至0~10℃,过滤,使用乙腈和水的混合液洗涤,滤饼于40~50℃,干燥,得盐酸帕洛诺司琼水合物晶型6.1g,收率为61.0%,纯度为99.78%,水含量为5.4%。
[0046]
实施例7盐酸帕洛诺司琼水合物晶型稳定性评价
[0047]
将制得的盐酸帕洛诺司琼晶体开展影响因素试验、加速稳定性试验,试验方法参见《中国药典(2015)》第二部附录xixc《原料药与药物制剂稳定性试验指导原则》。
[0048]
(一)影响因素试验:
[0049]
1.高温试验:取上述实施例中制备的盐酸帕洛诺司琼水合物晶型,于60℃放置10天,第10天取样,测定各指标与0天进行对照,试验结果见表1。
[0050]
2.高湿试验:取上述实施例中制备的盐酸帕洛诺司琼水合物晶型,于湿度在75%下放置10天,第10天取样,测定各指标与0天进行对照,试验结果见表1。
[0051]
3.强光照射试验:取上述实施例中制备的盐酸帕洛诺司琼,于照度为(4500
±
500)lux的条件下放置10天,第10天取样,测定各指标与0天进行对照,试验结果见表1。
[0052]
表1盐酸帕洛诺司琼水合物晶型影响因素试验
[0053][0054]
(二)加速稳定性试验:
[0055]
上述实施例中制备的盐酸帕洛诺司琼在恒温恒湿箱中进行1个月的加速稳定性试验。试验条件湿:40℃/75%相对湿度(rh),1个月后取样,进行纯度,有关物质和xrpd检测,结果见表2。
[0056]
表2盐酸帕洛诺司琼的加速稳定性试验
[0057]
试验条件时间纯度(%)有关物质(%)xrpd 0天99.8640.042
‑‑‑
40℃/75%(rh)1月99.8550.045未发生变化
[0058]
由上述影响因素试验和加速试验可以看出,本发明制备的盐酸帕洛诺司琼在高温、高湿和光照条件下杂质未见明显增加,纯度无明显变化,晶型未发生转变;加速稳定性试验证明产品质量稳定性良好。
[0059]
综上所述,本发明公开的盐酸帕洛诺司琼水合物晶型具有良好的稳定性,较低的引湿性、工艺可开发和易处理等有利性能。该水合物晶型的制备方法简单,成本低廉,易于工业化生产,对未来的药物的优化和开发具有重要价值。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1