改进的高效特异性的纤维蛋白溶解酶纤溶酶的人Kunitz抑制剂
本发明领域涉及能够抑制纤溶酶作用的多肽试剂。发明背景纤维蛋白溶解是一种生理过程,可调节凝块形成的程度及其在正常生理状况下的过度生长。然而,在外伤或外科手术、缺血和再灌注后,血液会接触到大的非内皮表面,如心肺旁路(cpb)回路,导致过度纤维蛋白溶解。此过度纤维蛋白溶解会导致凝血病、出血和炎症反应。在这些情况下,医务人员使用抑制纤溶酶(该过程的一种关键介质)的抗纤维蛋白溶解剂来减少出血、同种异体血液施用和不良的临床结果。使用的抗纤维蛋白溶解剂是抑肽酶、氨甲环酸(txa)和ε-氨基己酸(eaca)。抑肽酶在2008年从市场上撤出,因为主要由于肾功能不全和过敏反应在患者中其产生的不良反应(1)。值得注意的是,鉴于其风险效益概况,抑肽酶已以限制使用的方式重新引入欧洲和加拿大(2)。然而,美国并未解除对抑肽酶的禁令。目前使用的赖氨酸类似物txa和eaca不如抑肽酶有效,并且它们还会导致癫痫和肾功能不全(3,4)。蒙特利尔心脏外科小组最近发表的一篇文章(5)表明,2012年至2015年间,尽管几乎100%使用txa,但在该中心接受心脏外科手术的所有患者中,近四分之一的患者输注了超过4个单位的红细胞。这导致作者得出结论,仍然需要一种有效的血液保护剂。所需的产品必须限定针对纤维蛋白溶解和炎症的蛋白酶抑制概况,该概况具有收益与风险的最佳平衡(6)。近期,ecallantide和mdco-2010这两款抑制纤溶酶的产品完成了心脏搭桥手术研究直至ii期临床试验。然而,每种产品未能在iii期心脏搭桥手术试验中提供令人满意的疗效和安全性概况,并且两项试验都被提前终止(7,8)。这可能与ecallantide和mdco-2010是激肽释放酶的强抑制剂有关。此外,mdco-2010还抑制凝血蛋白酶因子xa。另一种产品,textilinin-1,是一种来自蛇毒的kunitz型丝氨酸蛋白酶抑制剂,其正处于开发的早期阶段(9)。在这种情况下,与来自牛肺的抑肽酶kunitz结构域相比,上述kunitz结构域来自蛇毒。因此,类似于抑肽酶,蛇毒kunitz结构域预计会在人中产生过敏反应。抑肽酶的问题以及该的技术人员为鉴定更适合体内使用的抗纤维蛋白溶解剂所做的持续努力表明,这是本领域公认的长期以来一直没有解决的问题,并且是本领域普通技术人员公认的持久需求。探索用作抗纤维蛋白溶解剂的一组试剂包括人kunitz型抑制剂多肽的变体(参见,例如美国专利号8,993,719和美国专利公开20080026998)。然而,由于这些人kunitz型抑制剂多肽可以在体内与包括纤溶酶和丝氨酸蛋白酶如激肽释放酶的多种多肽相互作用,这类多肽的复杂药代动力学对发现具有一系列为此类变体多肽提供经优化在体内用作治疗剂的功能特性的氨基酸残基的人kunitz型抑制剂变体多肽提出了挑战。
背景技术:
技术实现思路
1、如本文所公开的,已经制备了一种新的非天然存在的2型人组织因子途径抑制剂的kunitz结构域1(kd1)的多肽变体,并发现它具有非常理想的药代动力学概况。例如,所公开的多肽具有药代动力学概况,其包括比抑肽酶(一种常规使用但有问题的抗纤维蛋白溶解剂)更好地抑制纤溶酶活性的能力。此外,除了这种新的多肽变体的纤溶酶抑制活性优于用抑肽酶观察到的纤溶酶抑制活性之外,本文公开的多肽变体进一步避免了用抑肽酶和相关分子观察到的某些不良副作用。在一个示例中,观察到本文公开的多肽变体对其他凝血丝氨酸蛋白酶如激肽释放酶表现出极小的抑制活性。
2、本文公开的60个残基多肽变体包括独特的一组氨基酸残基,包括含有赖氨酸残基的c端结构。不受特定理论或作用机制的束缚,该c端结构看起来促进60个残基多肽变体以抑制纤溶酶原结合纤维蛋白凝块的方式通过其kringle结构域结合纤溶酶或纤溶酶原。本文公开的多肽变体还包括一组三个氨基酸突变(“kd1y11t/r15k/l17r-kt”),其包括第15位的赖氨酸氨基酸取代。令人惊讶的是,观察到与仅具有双重突变y11t/l17r的60个残基多肽变体相比,这种y11t/r15k/l17r三重突变体抑制纤溶酶的效力高4至5倍。不受特定理论或作用机制的束缚,这种在第15位具有赖氨酸氨基酸取代的三重突变体看起来通过促进这种变体多肽与纤溶酶中的残基asp189和ser 190的相互作用来发挥作用。出乎意料的是,与可比较的仅具有双重突变y11t/l17r的60个残基多肽变体相比,该60个残基y11t/r15k/l17r三重突变多肽进一步表现出至少弱10倍的对激肽释放酶、因子xia和因子viia/组织因子的抑制。因此,本文公开的60个残基变体多肽表现出非常理想的药代动力学/材料概况,包括例如强烈抑制纤溶酶的能力,同时避免与该技术中的类似抑制分子相关的某些副作用。
3、本文公开的发明具有多个实施方案。本发明的实施方案包括,例如包括包含以下序列的多肽的物质组合物:naeicllpldtgpckarllryyydrytqscrqflyggcegnannfytweacddacwriek(seq id no:1)和/或分离的非天然存在的多肽seq id no:1)。通常,此类物质组合物还包括额外的试剂,例如药学上可接受的载剂,如防腐剂、张力调节剂、去污剂、水凝胶、粘度调节剂、ph调节剂等。此类的实施方案包括,例如,药物组合物,其包括选择用于静脉内注射或输注的药学上可接受的赋形剂。
4、本发明的另一个实施方案是包括编码以下多肽序列的多核苷酸的物质组合物:naeicllpldtgpckarllryyydrytqscrqflyggcegnannf ytweacddacwriek(seq id no:1)。在本文公开的本发明的工作实施方案中,该多核苷酸包含以下序列:aacgcggagatctgtctcctgcccctagacaccggaccctgcaaagccagacttctccgttactactacgacaggtacacgcagagctgccgccagttcctgtacgggggctgcgagggcaacgccaacaatttctacacctgggaggcttgcgacgatgcttgctggaggatagaaaaa(seq id no:2)。本技术领域的技术人员理解,虽然该特定序列包含用于在人中产生本文公开的kunitz结构域1抑制剂多肽的密码子,但编码这些多肽的多核苷酸序列可以根据用于表达多肽的系统而变化(即,不同的密码子可以是用于细菌、酵母和昆虫细胞)。通常,此类多核苷酸位于包含一个或多个用于在细胞中表达多肽的调控序列的载体中。本发明的实施方案还包括包含此类载体的细胞(例如细菌、酵母、昆虫或哺乳动物细胞)。
5、如下所述,本发明的实施方案还包括使用本文公开的多肽的方法。本发明的此类实施方案包括,例如抑制纤溶酶的至少一种活性的方法,包括使纤溶酶与有效量的本文公开的多肽接触。本发明的相关实施方案包括抑制患者纤维蛋白溶解的方法,包括向患者施用足以抑制纤维蛋白溶解的量的本文公开的多肽,从而抑制纤维蛋白溶解。本发明的其他示例性实施方案包括治疗需要抑制纤溶酶活性的受试者的方法,所述方法包括向受试者施用有效量的本文公开的多肽。本发明的其他示例性实施方案包括治疗需要外科手术的受试者的方法,该方法包括在外科手术之前、期间和/或之后向受试者施用有效量的本文公开的多肽。本发明的其他示例性实施方案包括治疗患有癌症或癌前病况的受试者的方法,所述方法包括向受试者施用有效量的本文公开的多肽。本发明的其他示例性实施方案包括治疗受试者的可通过抑肽酶治疗的病况的方法,所述方法包括向受试者施用有效量的本文公开的多肽。
6、本发明的一个示例性实施方案是用于抑制受试者出血的方法,该方法包括向受试者施用有效量的本文公开的多肽。在本发明的某些实施方案中,出血由外科手术(例如,心脏外科手术或器官移植手术,如肝移植)或外伤(例如,外伤性脑损伤、枪伤、事故等)引起。本发明的一些实施方案包括其中配置有seq id no:1所示多肽的贴剂或敷布等。在这种情况下,本发明的另一个实施方案是抑制受试者体内纤溶酶的至少一种活性的方法,该方法通过将受试者的出血组织与其中配置有seq id no:1所示多肽的贴剂等接触,使得这些多肽能够抑制受试者中的纤溶酶的至少一种活性。
7、通过下面的详细描述,本领域技术人员将清楚本发明的其他目的、特征和优点。然而,应当理解,详细描述和具体实施例虽然指示了本发明的一些实施方案,但是以示例而非限制的方式给出的。在不脱离本发明的精神的情况下,可以在本发明的范围内进行许多改变和修改,并且本发明包括所有此类修改。
8、附图的简要说明
9、图1.来自纯化的kd1y11t/r15k/l17r-kt的sds-page凝胶电泳的数据。泳道1,蛋白质标记物;泳道2,还原的kd1y11t/r15k/l17r-kt;泳道3,非还原的kd1y11t/r15k/l17r-kt。
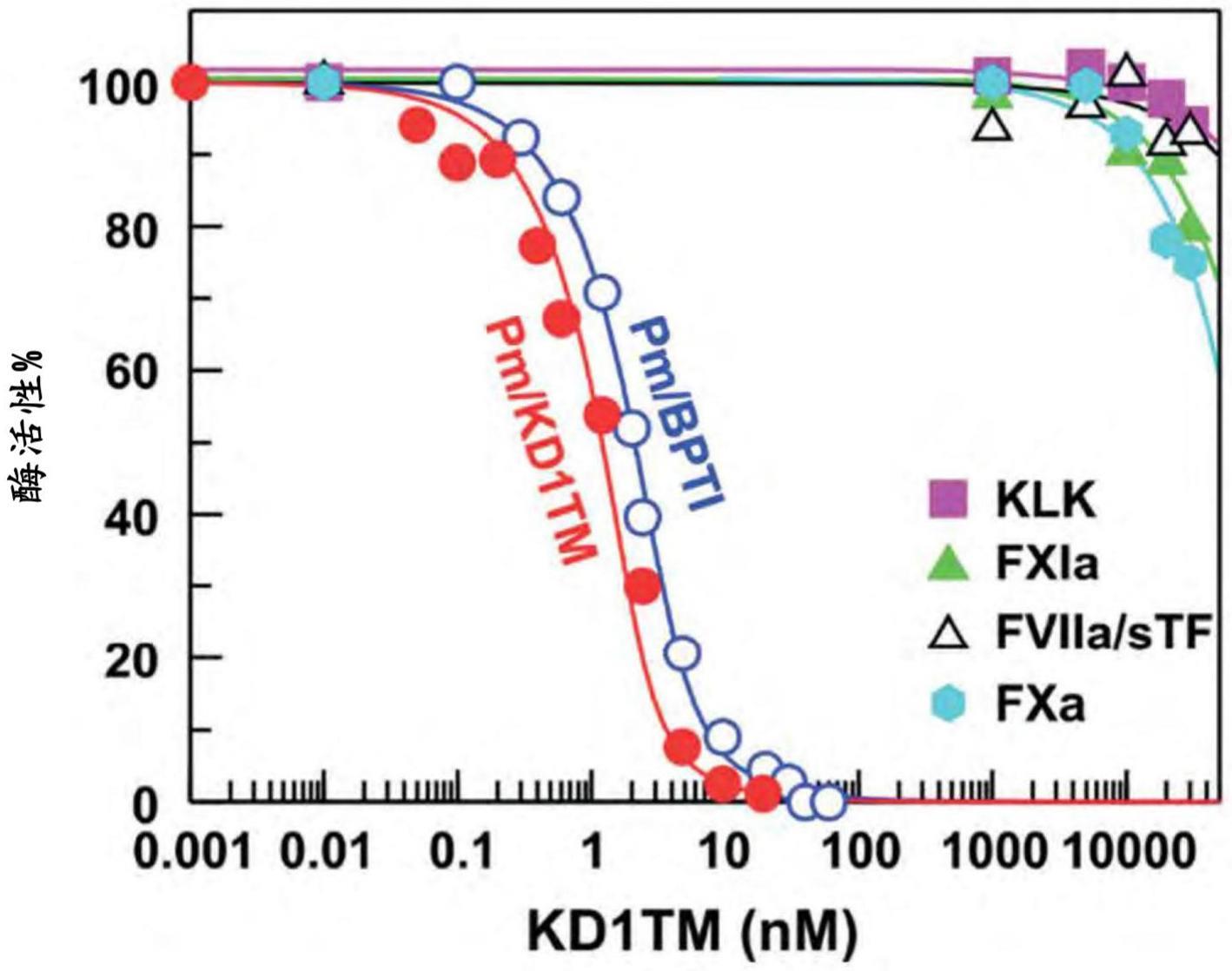
10、图2.来自kd1y11t/r15kl17r-kt与纤溶酶、fxia、激肽释放酶(klk)、fviia/stf和fxa的平徜抑制常数(ki)的数据。在不同浓度(0.05至19.2nm)的kd1y11t/r15kl17r-kt(●)或bpti(○)存在下,3nm人纤溶酶的剩余的活性百分比。以及在不同浓度(1μm至30μm)的kd1y11t/r15kl17r-kt存在下,1nm人fxia(▲)、1nm人kallikrein(■)、20nm人fviia/stf(δ)以及1nm的剩余的活性百分比。缩写:pm,纤溶酶;bpti,牛胰胰蛋白酶抑制剂(抑肽酶,特斯乐);kd1tm,kd1y11t/r15kl17r-kt;klk,激肽释放酶;fxia、因子xia、fviia/stf、因子viia/可溶性组织因子;fxa,因子xa。
11、图3a和3b.研究kd1y11t/r15kl17r-kt(a)或bpti(b)对人血浆中纤维蛋白溶解的影响的数据。将凝血酶(iia)添加到人血浆中以启动凝块形成,这与od405的增加有关(黑色;空心圆圈;iia,零tpa)。添加3μm的kd1y11t/r15kl17r-kt或bpti不影响凝块形成(棕色曲线;iia,3μm的kd1tm(图a)或bpti(图b))。添加tpa可将纤溶酶原转化为纤溶酶,纤溶酶在~12分钟内完全溶解纤维蛋白凝块,如od405最初增加随后减少所表示的(黑色;实心圆圈;iia,tpa)。添加kd]y11t/r15k/l17r-kt或bpti以剂量依赖性方式抑制纤维蛋白溶解--kd1y11t/r15k/l17r-kt(a)或bpti(b)如下:0.5μm(蓝色)、1μm(红色))、2μm(绿色)、3μm(洋红色)。缩写:tpa,组织纤溶酶原激活剂;kd1tm,kd1y11t/r15k/l17r-kt;bpti,牛胰胰蛋白酶抑制剂(抑肽酶/特斯乐)
12、图4.来自huvec生存力研究的数据。huvec未经处理或用指定浓度的以下抗纤维蛋白溶解剂处理24小时:kd1tm、bpti、eaca或txa。将细胞与细胞可渗透、无毒且弱荧光的蓝色指示剂染料刃天青一起孵育(alamarblue生存力测定)。荧光强度与相对细胞数成比例。误差条表示平均值的标准误差(sem)。发现每种抗纤维蛋白溶解剂处理的细胞的生存力与未处理的细胞没有显著差异(p>0.05)。缩写:kd1tm,kd1y11t/r15k/l17r-kt;bpti,牛胰胰蛋白酶抑制剂;eaca,ε-氨基己酸;txa,氨甲环酸。
13、图5.来自成纤维细胞生存力的研究的数据。成纤维细胞未经处理或用指定浓度的以下抗纤维蛋白溶解剂处理24小时:kd1tm、bpti、eaca或txa。将细胞与细胞可渗透、无毒且弱荧光的蓝色指示剂染料刃天青一起孵育(alamarblue生存力测定)。荧光强度与相对细胞数成比例。误差条表示平均值的标准误差(sem)。发现每种抗纤维蛋白溶解剂处理的细胞的生存力与未处理的细胞没有显著差异(p>0.05)。缩写:与上图4相同。
14、图6a和6b.来自huvec(a)和皮肤成纤维细胞(b)的细胞凋亡的研究的数据。huvec或成纤维细胞未经处理或用以下抗纤维蛋白溶解剂处理24小时:30μm的kd1tm、30μm的bpti、60mm的eaca、30mm的txa或0.05μm的紫杉醇(阳性对照)。显示为相对光单位(rlu)的发光与半胱天冬酶(caspase)-3/7活性成比例。误差条表示平均值的标准误差(sem)。缩写:kd1tm,kd1y11t/r15k/l17r-kt;bpti,牛胰胰蛋白酶抑制剂;eaca,ε-氨基己酸;txa,氨甲环酸;huvec,人脐静脉内皮细胞。
15、图7.来自huvec细胞毒性的研究的数据。huvec未经处理或用以下抗纤维蛋白溶解剂处理24小时:30μm的kd1tm、30μm的bpti、60mm的eaca、30mm的txa或0.05μm的紫杉醇作为阳性对照(上图)。fitc:当膜完整性受损时与dna结合的celltox绿色染料。荧光信号(在条形图中定量,底部图)与细胞毒性成比例。dapi:核染色剂,与所有细胞核结合。缩写:fitc,异硫氰酸荧光素;dapi,4′,6-二脒基-2-苯基吲哚;huvec,人脐静脉内皮细胞;kd1tm,kd1y11t/r15k/l17r-kt;bpti,牛胰胰蛋白酶抑制剂;eaca,ε-氨基己酸;txa,氨甲环酸。
16、图8.来自成纤维细胞细胞毒性的研究的数据。成纤维细胞未经处理或用以下抗纤维蛋白溶解剂处理24小时:30μm的kd1tm、30μm的bpti、60mm的eaca、30mm的txa或0.05μm的紫杉醇以及溶解缓冲液作为阳性对照(上图)。fitc:当膜完整性受损时,与dna结合的celltox绿色染料。荧光信号(在条形图中定量,底部图)与细胞毒性成比例。dapi:核染色剂,与所有细胞核结合。缩写:fitc,异硫氰酸荧光素;dapi,4′,6-二脒基-2-苯基吲哚;kd1tm,kd1y11t/r15k/l17r-kt;bpti,牛胰胰蛋白酶抑制剂;eaca,ε-氨基己酸;txa,氨甲环酸。
17、图9a-9b.人组织因子途径抑制剂的kunitz结构域1(kd1)的模拟复合物。图9a显示了kd1y11t/r15k/l17r-kt与纤溶酶相互作用的模拟复合物。(a)kd1y11t/r15k/l17r-kt与纤溶酶蛋白酶结构域的模拟相互作用。描绘了纤溶酶蛋白酶结构域的静电表面和kd1y11t/r15k/l17r-kt(浅绿色)的卡通表示。kd1y11t/r15k/l17r-kt的p1(lys15)、p5(thrll)和p2′(arg17)残基与纤溶酶相互作用以棒状表示形式显示。在静电表面,蓝色代表正电荷,红色代表负电荷,白色代表中性电荷。(b)kd1y11t/r15k/l17r-kt与纤溶酶kringle结构域的模拟相互作用。描述了纤溶酶原kringle结构域1的静电表面和kd1y11t/r15k/l17r-kt(浅绿色)的卡通表示。在kringle结构域和kd1y11t/r15k/l17r-kt之间形成氢键和盐桥(显示为虚线)的残基以棒状表示形式显示。kringle结构域的碳原子显示为绿色,kd1y11t/r15k/l17r-kt的碳原子显示为浅绿色。如(a)中所示,氧原子以红色显示,氮原子以蓝色显示。kd1y11t/r15k/l17r-kt残基用后缀i标记。在静电表面,蓝色代表正电荷,红色代表负电荷,白色代表中性电荷。图9b显示kd1-y11t/l17r-kt与纤溶酶相互作用的模拟复合物。(a)kd1-y11t/l17r-kt与纤溶酶蛋白酶结构域的模拟相互作用。描绘了纤溶酶蛋白酶结构域的静电表面和kd1-y11t/l17r-kt(黄色)的卡通表示。kd1-y11t/l17r-kt的p1(arg15)、p5(thr11)和p2′(arg17)残基与纤溶酶相互作用以棒状表示形式显示。在静电表面,蓝色代表正电荷,红色代表负电荷,白色代表中性电荷。(b)kd1-y11t/l17r-kt与纤溶酶kringle结构域的模拟相互作用。描述了纤溶酶原kringle结构域1的静电表面和kd1-y11t/l17r-kt(黄色)的卡通表示。在kringle结构域和kd1-y11t/l17r-kt之间形成氢键和盐桥(显示为虚线)的残基以棒状表示形式显示。kringle结构域的碳原子显示为绿色,kd1-y11t/l17r-kt的碳原子显示为黄色。如(a)中所示,氧原子以红色显示,氮原子以蓝色显示。kd1-y11t/l17r-kt残基用后缀i标记。在静电表面,蓝色代表正电荷,红色代表负电荷,白色代表中性电荷。
18、图10.kd1多肽的序列。人tfpi-2 kd1单突变体(kd1-l17r-kt,)在大肠杆菌中的表达序列(seq id no:4)和大肠杆菌中的双突变体(kd1-y11t/l17r-kt)(seq id no:5)和巴斯德毕赤酵母中的双突变体(kd1-y11t/l17r-kt)(seq id no:6)。向下箭头表示为去除his标签而引入的肠激酶切割位点。突变的残基tyr11thr和leu17arg也被标记。残基1根据bpti-kunitz结构域编号进行编号,对应于tfpi-2kunitz结构域1序列中的氨基酸10。
19、图11.来自tfpi-2kd1单突变体和双突变体的sds-page分析研究的数据。泳道1,分子量(mw)标记物;泳道2,还原的大肠杆菌kd1-l17r-kt;泳道3,还原的大肠杆菌kd1-y11t/l17r-kt;泳道4,还原的巴斯德毕赤酵母kd1-y11t/l17r-kt;泳道5,非还原的大肠杆菌kd1-l17r-kt;泳道6,非还原的大肠杆菌kd1-y11t/l17r-kt;泳道7,非还原的巴斯德毕赤酵母kd1-y11t/l17r-kt。每条泳道上样了5μg蛋白质。kd1sm,kd1-l17r-kt;和kd1dm、kd1-y11t/l17r-kt。
20、图12a和12b.来自以下研究的数据:12(a)大肠杆菌表达的kd1-wt(iia切割位点,32),大肠杆菌表达的kd1-l17r-kt,有肠激酶切割位点的kd1-y11t/l17r-kt,巴斯德毕赤酵母表达的kd1-y11t/l17r-kt和抑肽酶与纤溶酶的平衡解离常数(ki)的确定。酶活性表示为在增加的抑制剂浓度下的分数活性百分比(抑制率/未抑制率)。抑制常数(ki)使用方程1和2确定,如实验部分所述。数据代表三个实验的平均值。使用的纤溶酶浓度为3nm。12(b)kd1-y11t/l17r-kt与fviia/stf、pklk和fxia的抑制概况。fviia/stf的浓度为20nm,而pklk和fxia各为1nm。在进行的一式三份实验中,kd1-y11t/l17r-kt浓度高达3μm时未观察到抑制作用。
21、图13a和13b。来自通过spr测量的kd1-y11t/l17r-kt与dip-δ纤溶酶和tpa的相互作用的研究的数据。(a)dip-δ纤溶酶与kd1-y11t/l17r-kt结合。dip-δ纤溶酶通过胺偶联法偶联到cm5芯片上,结合蛋白的固定化水平达到734个反应单位(ru)。使用了五个浓度(0.1μm、0.3μm、0.5μm、0.75μm和1μm)的kd1-y11t/l17r-kt,结合时间为6分钟,解离时间为10分钟(流速为10μl/min)。详细信息在实验部分提供。(b)tpa与kd1-y11t/l17r-kt的结合。tpa与cm5芯片偶联,结合蛋白的固定水平达到1182ru。使用了五个浓度(0.1μm、0.3μm、0.75μm、1μm和2μm)的kd1-y11t/l17r-kt。分析物结合和解离方案与图(a)中的相同。图a和图b中的实验一式两份进行。然后使用每个数据集计算kon、koff和kd值,并获得文中提供的平均值±sd值。
22、图14a-14c.来自kd1-l17r-kt、kd1-y11t/l17r-kt和抑肽酶对人npp中纤维蛋白溶解的影响的研究的数据。将iia添加到npp以启动凝块形成,这与od405的增加有关(曲线o;a-c中的iia,零tpa)。同时添加tpa将纤溶酶原转化为纤溶酶,纤溶酶在约10min内完全溶解了纤维蛋白凝块,如od405的初始增加和随后的减少所示(曲线●;a-c中的iia,tpa)。添加kd1-l17r-kt、kd1-y11t/l17r-kt或抑肽酶以剂量依赖性方式抑制纤维蛋白溶解。(a)kd1-l17r-kt的影响;0.5μm(■)、1μm(□)、1.5μm(▲)、4μm(■)和5μm(□)。(b)kd1-y11t/l17r-kt的影响;0.5μm(■)、1μm(■)、1.5μm(▲)、2μm(▲)和(c)抑肽酶的作用;0.5μm(■)、1μm(□)、1.5μm(▲)、2μm(△)和
23、图15a-15d.来自在血浆凝块溶解测定中使用的不同浓度下的kd1-l17r-kt、kd1-y11t/l17r-kt和抑肽酶的纤维蛋白溶解中点的比较研究的数据。条形图是用指定浓度的kd1-l17r-kt、kd1-y1 1t/l17r-kt和抑肽酶达到纤维蛋白溶解中点的呈现显示时间(分钟)。每个图都标明了所用每种抑制剂的浓度。所有实验一式三份进行,并显示了平均值±sd值。注:不带*的条形表示与列出的所有其他试剂有显著差异。*表示p<0.05。
24、图16a-16g.来自血栓弹性成像的数据显示了kd1-wt、kd1-l17r-kt、kd1-y11t/l17r-kt、抑肽酶和eaca的剂量反应分析。所有实验均包含柠檬酸化全血(300μμl)、1.5μm纤溶酶和10mm cacl2。首先将抗纤维蛋白溶解剂添加到血液中,然后掺入1.5μm纤溶酶和10mmcacl2。监测凝块形成和溶解180分钟。在没有任何抗纤维蛋白溶解剂的情况下,在存在或不存在纤溶酶的情况下进行对照实验。(a)凝块形成和纤维蛋白溶解的纤溶酶作用。向柠檬酸化全血(300μl)中掺入各种浓度的纤溶酶(0-3μm)和10mm cacl2。监测凝块形成和溶解180分钟。1μm(b)、2μm(c)、3μm(d)、4μm(e)、5μm(f)和7.5μm(g)的kd1-wt、kd1-l17r-kt、kd1-y11t/l17r-kt和抑肽酶对凝块形成和使用1.5μm的纤溶酶的纤维蛋白溶解的影响。(g)200μm至3000μm的eaca对凝块形成和使用1.5m的纤溶酶的纤维蛋白溶解的影响。pm,纤溶酶;nhb,正常人血。
25、图17a-17f.来自不同浓度下kdi-wt、kd1-l17r-kt、kd1-y11t/l17r-kt、抑肽酶和eaca的teg实验的最大幅度(ma)的比较研究的数据。条形图表示用不同浓度的kd1-wt、kd1-l17r-kt、kd1-y11t/l17r-kt、抑肽酶和eaca获得的ma——a,1μm;b,2μm,c,3μm;d,5μm,e,7.5μm和f,eaca。所有实验一式两份进行,并显示了平均值±sd值。注:不带*的条形表示与列出的所有其他试剂有显著差异。*表示p<0.05。
26、图18a-18f.来自不同浓度的kd1-wt、kd1-l17r-kt、kd1-y11t/l17r-kt、抑肽酶和eaca的剪切弹性模量强度(g、teg实验)的比较研究的数据。条形图表示用不同浓度的kd1-wt、kd1-l17r-kt、kd1-y11t/l17r-kt、抑肽酶和eaca获得的“g”-a,1μm;b,2μm,c,3μm;d,5μm,e,7.5μm和f,eaca。所有实验一式两份进行,并呈现了平均值±sd值。注:不带*的条形表示与列出的所有其他试剂有显著差异。*表示p<0.05。
27、图19a-19d.来自不同浓度的kd1-wt、kd1-l17r-kt、kd1-y11t/l17r-kt和抑肽酶的ly60%(teg实验)比较研究的数据。描绘了显示60分钟时溶解百分比的条形图。所有实验一式两份进行,并显示了平均值±sd值。注:不带*的条形表示与列出的所有其他试剂有显著差异。*表示p<0.05。
28、图20.说明kd1tm剂量反应分析的血栓弹性成像。所有实验均包含柠檬酸化全血(300μl)、0.15μm凝血酶和10mm cacl2。首先将kd1tm添加到血液中,然后掺入0.15μm凝血酶、2nmtpa和10mm cacl2。监测凝块形成和溶解180分钟。在没有kd1tm的情况下,在存在或不存在2nm tpa的情况下进行对照实验(曲线1和曲线2)。使用0.15μm α-凝血酶和2nm tpa的情况下,kd1tm对凝块形成和纤维蛋白溶解的影响(曲线3、2μm和曲线4、4μm)。
29、发明详述
30、在对实施方案的描述中,可以参考构成其一部分的附图,并且在附图中以示例的方式示出了可以实践本发明的特定实施方案。应当理解,可以使用其他实施方案,并且可以在不脱离本发明的范围的情况下进行结构改变。除非另有定义,本文使用的所有技术术语、符号和其他科学术语或术语旨在具有本发明所属领域的技术人员通常理解的含义。在一些情况下,为了清楚和/或便于参考,本文定义了具有通常理解的含义的术语,并且本文中包含的此类定义不一定被解释为表示与本领域通常理解的内容存在实质性差异。本文描述或引用的技术和过程的许多方面为本领域技术人员所熟知和普遍采用。下文讨论了本发明的各种实施方案。
31、抗纤维蛋白溶解剂,例如所公开的多肽,可用于减少主要外科手术和外伤,如心脏外科手术、骨科手术、肝脏手术、神经外科和产科期间的出血和输血。如本文所公开的,已经制备了具有序列naeicllpldtgpckarllryyydrytqscrqflyggcegnannfytweacddacwriek(seq id no:1)的2型人组织因子途径抑制剂的kunitz结构域1(kd1)的新多肽变体,并发现其具有对体内使用非常需要的药代动力学概况。本文公开的这种60个残基多肽变体包括独特的氨基酸残基群,包括含有赖氨酸残基的c端结构。本文公开的多肽变体进一步包括一组三个氨基酸突变(“kd1y11t/r15k/l17r”),包括第15位的赖氨酸氨基酸取代。令人惊讶的是,观察到该y11t/r15k/l17r三重突变体抑制纤溶酶的效力比仅具有双重突变y11t/l17r的60个残基多肽变体高4至5倍。令人惊讶的是,与仅具有双重突变y11t/l17r的可比较的60个残基多肽变体相比,该60个残基y11t/r15k/l17r三重突变体多肽进一步表现出对激肽释放酶、因子xia和因子viia/组织因子至少弱10倍的抑制。因此,本文公开的60个残基变体多肽表现出高度理想的药代动力学/材料概况,包括例如,强烈抑制纤溶酶的能力,同时避免与该技术中的类似抑制分子(例如,抑肽酶)相关的某些副作用。因此,本文所公开的60个残基变体多肽满足了长期存在的需求,该需求已被认可、其持续存在且未被其他人解决。
32、本文公开的发明具有多个实施方案。本发明的实施方案包括例如,包括包含序列naeicllpldtgpckarllryyydrytqscrqflyggcegnannfytweacddacwriek(seq id no:1)(或基本上由以下序列组成)的多肽的物质组合物。通常,此类物质组合物还包括额外的试剂,例如药学上可接受的载剂,如防腐剂、张力调节剂、去污剂、水凝胶、粘度调节剂、ph调节剂等。本发明的此类实施方案包括,例如,药学上可接受的组合物,其包含60个氨基酸的蛋白质序列,该蛋白质序列表示为:naeicllpldtgpckarllryyydrytqscrqflyggcegnannfytweacddacwriek(seq id no:1);以及适于静脉内注射或输注的药学上可接受的赋形剂。此类药学上可接受的赋形剂在本领域中是众所周知的,并且在remington′s pharmaceuticalsciences(mack pub.co.,nj current edition)中对药学上可接受的载剂、稀释剂和其他赋形剂进行了详尽的讨论。
33、此外,本文公开的2型人组织因子途径抑制剂的kunitz结构域1(kd1)的多肽变体进一步表现出理想的和意想不到的稳定性概况。例如,在本发明的某些实施方案中,其中当该多肽组合物在包含0.1mg/ml的牛血清白蛋白(bsa)和2mm钙的tris缓冲盐水(tbs)中在37℃孵育至少2天、4天或1周时,所述多肽的纤溶酶抑制常数(ki)变化小于10%(或小于5%)。参见,例如下面实施例2中的表7。
34、本发明的实施方案包括本文公开的多肽组合物,其进一步配置在贴剂或敷布材料等的基底内。如本领域已知的,此类贴剂或敷布材料可用于实现流血伤口的止血。在本发明的上下文中,基底被理解为包括任何类型的医用敷布、贴剂、海绵、垫、拭子、敷料等,因为它们通常用于医疗领域的伤口处理。基底例如可以由棉和/或纤维素基材料(粘胶或人造丝)制成,例如以吸收性织造或非织造纺织品的形式。在上下文中,本发明的一个此类实施方案是贴剂、海绵、垫、拭子、敷料等,其具有配置在贴剂、海绵、垫、拭子、敷料等的基质内的本文公开的60个氨基酸的多肽(如seq id no:1所示)。在典型的实施方案中,贴剂等被设计成配置在体内的位置(例如,损伤位点)并且60个氨基酸的多肽配置在贴剂内,使得该多肽可以扩散远离贴剂等的基质进入贴剂配置位点的周围组织。可以适用于本发明的此类实施方案的示例性贴剂和类似材料和方法在例如美国专利公开号20020049471、20110071498和20210038758中公开,其内容通过引用并入。
35、本发明的另一个实施方案是包括编码多肽序列naeicllpldtgpckarllryyydrytqscrqflyggcegnannfytweacddacwriek(seq id no:1)的多核苷酸的物质组合物。在本文公开的本发明的工作实施方案中,该多核苷酸包含序列aacgcggagatctgtctcctgcccctagacaccggaccctgcaaagccagacttctccgttactactacgacaggtacacgcagagctgccgccagttcctgtacgggggctgcgagggcaacgccaacaatttctacacctgggaggcttgcgacgatgcttgctggaggatagaaaaa(seqid no:2)。本技术领域的技术人员理解,虽然该特定序列包含用于在人中产生本文公开的kunitz结构域1抑制剂多肽的密码子,但编码这些多肽的多核苷酸序列可以根据用于表达多肽的系统而变化(即,在细菌、酵母和昆虫细胞中可以使用不同的密码子)。通常,此类多核苷酸放置于包含一个或多个用于在细胞中表达多肽的调控序列的载体中。本发明的实施方案还包括包含此类载体的细胞(例如,细菌、酵母、昆虫或哺乳动物细胞)。
36、本发明的实施方案还包括使用本文公开的多肽的方法。本发明的此类实施方案包括,例如,抑制患者纤维蛋白溶解的方法,包括向患者施用足以抑制纤维蛋白溶解的量的本文公开的多肽,从而抑制纤维蛋白溶解。本发明的相关实施方案包括用于抑制纤溶酶的至少一种活性的方法,该方法包括使纤溶酶与有效量的本文公开的多肽接触。本发明的其他示例性实施方案包括治疗受试者的可通过抑肽酶治疗的病况的方法,所述方法包括向受试者施用有效量的本文公开的多肽。本发明的其他示例性实施方案包括治疗需要抑制纤溶酶活性的受试者的方法,所述方法包括向受试者施用有效量的本文公开的多肽。本发明的其他示例性实施方案包括治疗需要外科手术的受试者的方法,该方法包括在外科手术之前、期间和/或之后向受试者施用有效量的本文公开的多肽。本发明的其他示例性实施方案包括治疗患有癌症或癌前病况的受试者的方法,所述方法包括向受试者施用有效量的本文公开的多肽。本发明的一个此类的示例性实施方案包括治疗患有癌症转移的受试者的方法,所述方法包括向受试者施用有效量的本文公开的多肽。
37、本发明的相关实施方案包括用于抑制受试者出血的方法,所述方法包括向受试者施用有效量的本文公开的多肽。在本发明的某些实施方案中,出血由裂伤(例如,肝裂伤)、外科手术(例如,器官如肝移植)或外伤(例如,外伤性脑损伤、枪伤、事故等)引起。在本发明的这些方法的某些中,将多肽递送至贴剂中的体内位置,该贴剂中配置于其中的60个氨基酸的多肽。在这种情况下,本发明的另一个实施方案是用于抑制受试者出血的方法,该方法包括使贴剂与出血组织接触,使得60个氨基酸的多肽可以从贴剂基质扩散开并进入体内环境(例如,出血组织)。例如,本文提供治疗患有例如过度出血的患者的方法,该方法包括向患者施用治疗有效量的所公开的多肽。在其他实施方案中,本文提供治疗患有例如,中风(例如,急性中风)、由中风引起的脑缺血、脑血肿、脑水肿、乏氧/缺氧性脑损伤或外伤性脑损伤的患者的方法,该方法包括向患者施用治疗有效量的公开的多肽。
38、本发明的一个示例性实施方案是治疗正在接受心脏外科手术并需要减少失血的患者的方法,该方法包括外科手术之前、外科手术期间或外科手术之后施用治疗有效量的药学上可接受的组合物以及适于静脉内注射或输注的药学上可接受的赋形剂,该组合物包含由naeicllpldtgpckarllryyydrytqscrqflyggcegnannfytweacddacwriek seq id no:1)所示的60个氨基酸的蛋白质序列。在本发明的某些实施方案中,心脏外科手术是心肺旁路手术。本发明的另一个实施方案是治疗创伤性失血性休克的患者的方法,该方法包括外科手术之前、外科手术期间或外科手术之后施用治疗有效量的药学上可接受的组合物以及适于的静脉内注射或输注的药学上可接受的赋形剂,该组合物包含由(seq id no:1)所示的60个氨基酸的蛋白质序列。此外,由于rhukd1-tm的强大的纤溶酶抑制特性(且无抗凝特性),它可以以类似于过去使用抑肽酶治疗血友病的方式用于预防性治疗血友病。此外,本文公开的多肽进一步具有优于抑肽酶的优点。在一个这样的实例中,由于观察到使用抑肽酶(牛源性)会发生过敏反应,因此在此类治疗方案中不能长时间使用抑肽酶。
39、本发明的实施方案包括本文公开的2型人组织因子途径抑制剂的kunitz结构域1(kd1)的60个氨基酸多肽变体的给药方法。例如,本发明的实施方案包括在治疗方法中(例如,在抑制患者纤溶酶的至少一种活性的方法中)施用该60个氨基酸多肽变体的剂量为约1微克多肽变体每克患者体重至约10微克多肽变体每克患者体重(例如,约2至约8微克多肽变体每克患者体重,约4微克多肽变体每克患者体重等)。此外,本发明的实施方案包括专门设计用于治疗急性和/或慢性医学病况的给药/施用方法。例如,在本发明的某些实施方案中,60个氨基酸多肽变体用于选择在外科手术/损伤之后不久,例如外科手术/受伤后小于48小时、小于24小时、小于12小时或小于4小时施用该多肽变体的方法。在本发明的一些实施方案中,60个氨基酸多肽变体用于选择在外科手术损伤后较长时间段,例如外科手术/受伤后至少2天、4天、7天、14天或21天施用该多肽变体的方法。
40、多肽和相关方法的详细方面和实施方案在下面的实施例中公开。本发明的额外方面和实施方案在以下部分中讨论。
41、本发明的方面和实施方案
42、在本文所公开的发明的上下文中,术语“多肽”、“蛋白质”和“肽”和“糖蛋白”可互换使用并且意指不限于任何特定长度的氨基酸聚合物。该术语不排除诸如十四烷基化、硫酸化、糖基化、磷酸化、甲酰化以及信号序列的添加或缺失的修饰。术语“多肽”或“蛋白质”是指一条或多条氨基酸链,其中每条链包含通过肽键共价连接的氨基酸,并且其中所述多肽或蛋白质可包含多条通过肽键非共价和/或共价连接在一起的链,其具有天然蛋白的序列,即由天然存在的和特别是非重组细胞或基因工程化或重组细胞产生的蛋白质,并包含具有天然蛋白氨基酸序列的分子,或具有天然序列的一个或多个氨基酸的缺失、添加和/或取代的分子。因此,“多肽”或“蛋白质”可包含一个(称为“单体”)或多个(称为“多聚体”)氨基酸链。术语“肽”、“多肽”和“蛋白质”具体涵盖本公开的免疫调节多肽,或具有免疫调节多肽的一个或多个氨基酸的缺失、添加和/或取代的序列。
43、术语“分离的”是指材料从其原始环境(例如,如果是天然存在的,则为自然环境)移除。例如,存在于活动物中的天然存在的多肽或核酸不是分离的,但是从自然系统中的一些或所有共存物质分离的相同多肽或核酸是分离的。此类核酸可以是载体的一部分和/或此类核酸或多肽可以是组合物(例如,细胞溶解物)的一部分,并且仍然是分离的,因为此类载体或组合物不是核酸或多肽的自然环境的一部分。术语“基因”是指涉及产生多肽链的dna的区段;它包括编码区“前导和尾部”前后的区域以及各个编码区段(外显子)之间的间插序列(内含子)。
44、本文中提及的术语“分离的蛋白”和“分离的多肽”是指主题蛋白或多肽(1)不含至少一些通常在自然界中发现的其他蛋白质或多肽,(2)基本上不含来自相同来源,例如来自相同物种的其他蛋白质或多肽,(3)由来自不同物种的细胞表达,(4)已与至少约50%的多核苷酸、脂质、碳水化合物或与它在自然界中缔合的其他材料分离,(5)与“分离蛋白”或“分离多肽”在自然界中可能与之缔合的蛋白质或多肽部分不缔合(通过共价或非共价相互作用),(6)与在自然界中不与其缔合的多肽可操作地缔合(通过共价或非共价相互作用)或(7)在自然界中不存在。此类分离的蛋白或多肽可以由基因组dna、cdna、mrna或其他rna编码,或它们可以来自于根据许多用于人工肽和蛋白合成的众所周知的化学中的任何一种或其任何组合的合成来源。在某些实施方案中,分离的蛋白或多肽基本上不含在其自然环境中发现的会干扰其使用(治疗、诊断、预防、研究或其他)的蛋白质或多肽或其他污染物。
45、某些实施方案涉及编码本文所述的纤溶酶抑制多肽的核酸分子。用于产生期望核酸和/或多肽的方法是本领域众所周知的。例如,核酸和/或多肽可以从细胞中分离或通过化学合成从头合成。此类核酸或多肽可掺入载体中,并转化入宿主细胞中。宿主细胞可以在标准营养培养基中培养,加上必要的补充物或添加剂以诱导启动子、选择转化体或扩增适当的序列。
46、此外,编码纤溶酶抑制多肽的多核苷酸或多肽变体可分别含有相对于天然(例如,野生型或主要或天然存在的等位基因形式)的一个或多个核苷酸或氨基酸取代、添加、缺失和/或插入。在一些实施方案中,变体包括其中n端l-氨基酸被d-氨基酸替代的分子,并且在某些其他实施方案中,一个或多个其他氨基酸(例如,不位于n端)可以另外或替代地用d-氨基酸替代。在某些实施方案中,变体包括其中n端α氨基酸被β或γ氨基酸替代的分子。变体与纤溶酶抑制多肽序列或编码此类多肽的多核苷酸序列的一部分优选表现出至少约75%、78%、80%、85%、87%、88%或89%的同一性,并且更优选至少约90%、91%、92%、93%、94%、95%、96%、97%、98%或99%的同一性。通过将多肽或多核苷酸变体的序列与全长多核苷酸或多肽的相应部分进行比较,可以很容易地确定同一性百分比。用于序列比较的一些技术包括使用本领域普通技术人员熟知的计算机算法,诸如align或blast算法(altschul,j.mol.biol.219:555-565,1991;henikoffff and henikoff,pnas usa 89:10915-10919,1992))。可以使用默认参数。
47、此外,本文公开的纤溶酶抑制多肽变体可以偶联至额外的分子或试剂,诸如显像剂、颗粒、聚合物或其他试剂,包括促进多肽递送至特定体内位置的那些(例如抗体、肽等)。本发明的此类实施方案包括2型人组织因子途径抑制剂的kunitz结构域1(kd1)的多肽变体,其包含以下序列(或基本上由其组成):naeicllpldtgpckarllryyydrytqscrqflyggcegnannfytweacddacwriek(seq id no:1),其与额外的分子或试剂偶联。在一个这样的实例中,本发明的实施方案可以以这种方式进行修饰,以促进多肽向cns的递送(参见,例如behzad et al.,(2019),expert opinion on drug delivery,16∶6,583-605;salameh etal.,advpharmacol.2014;71:277-99;和美国专利公开号20200230218,20060189515,20150174267和20160213760,其内容通过引用并入)。
48、术语“可操作地连接”是指应用该术语的组分处于允许它们在合适的条件下执行其固有功能的关系中。例如,将与蛋白质编码序列“可操作地连接”的转录控制序列连接到其上,以便在与控制序列的转录活性相容的条件下实现蛋白质编码序列的表达。
49、如本文所用,术语“控制序列”是指可影响与其连接或可操作连接的编码序列的表达、加工或细胞内定位的多核苷酸序列。此类控制序列的本质可能取决于宿主生物体。在具体实施方案中,原核生物的转录控制序列可包括启动子、核糖体结合位点和转录终止序列。在其他具体实施方案中,真核生物的转录控制序列可包括含有一个或多个转录因子识别位点的启动子、转录增强子序列、转录终止序列和聚腺苷酸化序列。在某些实施方案中,“控制序列”可以包括前导序列和/或融合伴侣序列。
50、本文所指的术语“多核苷酸”是指单链或双链核酸聚合物。在某些实施方案中,包含多核苷酸的核苷酸可以是核糖核苷酸或脱氧核糖核苷酸或任一类型核苷酸的修饰形式。此类修饰可包括碱基修饰诸如溴尿苷、核糖修饰诸如阿拉伯糖苷和2′,3′-双脱氧核糖和核苷酸间联接修饰诸如硫代磷酸酯、二硫代磷酸酯、硒代磷酸酯、二硒代磷酸酯、苯胺硫代磷酸酯(phosphoroanilothioate)、苯胺磷酸酯和氨基磷酸酯。术语“多核苷酸”具体包括单链和双链形式的dna。
51、术语“天然存在的核苷酸”包括脱氧核糖核苷酸和核糖核苷酸。术语“修饰的核苷酸”包括具有修饰或取代的糖基等的核苷酸。术语“寡核苷酸联接”包括寡核苷酸联接,诸如硫代磷酸酯、二硫代磷酸酯、硒代磷酸酯、二硒代磷酸酯、苯胺硫代磷酸酯、苯胺磷酸酯、氨基磷酸酯等。参见,例如,laplanche et al.,1986,nucl.acids res.,14:9081;stec etal.,1984,j.am.chem.soc.,106:6077;stein et al.,1988,nucl.acids res.,16:3209;zon et al.,1991,anti-cancer drug design,6:539;zon et al.,1991,oligonucleotides and analogues:a practical approach,pp.87-108(f.eckstein,ed.),oxford university press,oxford england;stec et al.,u.s.pat.no.5,151,510;uhlmann and peyman,1990,chemical reviews,90:543,出于任何目的,其公开内容通过引用并入本文。寡核苷酸可以包括可检测标记以能够检测寡核苷酸或其杂交。
52、术语“载体”用于指代用于将编码信息转移至宿主细胞的任何分子(例如,核酸、质粒或病毒)。术语“表达载体”是指适合于转化宿主细胞并含有指导和/或控制插入的异源核酸序列表达的核酸序列的载体。如果存在内含子,表达包括但不限于诸如转录、翻译和rna剪接的过程。
53、如本领域技术人员所理解的,多核苷酸可包括基因组序列、基因组外和质粒编码的序列和较小的工程化基因区段,其表达或可适于表达蛋白质、多肽、肽等。此类区段可以是天然分离的,或由技术人员合成修饰的。
54、技术人员还将认识到,多核苷酸可以是单链的(编码或反义)或双链的,并且可以是dna(基因组、cdna或合成的)或rna分子。rna分子可包括含有内含子并以一对一方式对应于dna分子的hnrna分子,以及不含有内含子的mrna分子。根据本公开,额外的编码或非编码序列可以但不必存在于多核苷酸内,并且多核苷酸可以但不必连接至其他分子和/或支持材料。多核苷酸可包含天然序列或可包含编码此类序列的变体或衍生物的序列。
55、因此,根据这些和相关的实施方案,本公开还提供了编码本文所述的纤溶酶抑制多肽的多核苷酸。在某些实施方案中,提供的多核苷酸包含编码如本文所述的纤溶酶抑制多肽的一些或所有多核苷酸序列,以及此类多核苷酸的互补物。
56、在其他相关实施方案中,多核苷酸变体可与编码本文所述的纤溶酶抑制多肽的多核苷酸序列具有实质同一性。例如,多核苷酸可以是使用本文描述的方法(例如,使用标准参数的blast分析,如下所述),与参考多核苷酸序列,诸如编码具有本文公开的氨基酸序列的纤溶酶抑制多肽的序列相比包含至少70%序列同一性,优选至少75%、80%、85%、90%、95%、96%、97%、98%或99%或更高序列同一性的多核苷酸。本领域技术人员将认识到,通过考虑密码子简并性、氨基酸相似性、阅读框定位等,可以适当地调整这些值以确定由两个核苷酸序列编码的蛋白质的相应同一性。
57、通常,多核苷酸变体将含有一个或多个取代、添加、缺失和/或插入,优选地使得由变体多核苷酸编码的纤溶酶抑制多肽对纤溶酶的结合亲和力相对于具有本文具体列出的氨基酸序列的纤溶酶抑制多肽的结合亲和力不显著降低。
58、根据某些相关实施方案,提供了重组宿主细胞,其包含一种或多种如本文所述的构建体;编码纤溶酶抑制多肽或其变体的核酸;和产生所编码产物的方法,该方法包括从其编码核酸表达。通过在合适的条件下培养含有核酸的重组宿主细胞可以方便地实现表达。在通过表达产生后,可以使用任何合适的技术分离和/或纯化纤溶酶抑制多肽,然后按期望使用。
59、用于在多种不同宿主细胞中克隆和表达多肽的系统是众所周知的。合适的宿主细胞包括细菌、哺乳动物细胞、酵母和杆状病毒系统。本领域可用于表达异源多肽的哺乳动物细胞系包括中国仓鼠卵巢细胞、hela细胞、幼仓鼠肾细胞、nso小鼠黑色素瘤细胞和许多其他细胞。常见的、优选的细菌宿主是大肠杆菌。
60、肽在原核细胞诸如大肠杆菌中的表达在本领域中是公认的。综述参见例如pluckthun,a.bio/technology 9:545-551(1991)。作为生产重组多肽的选择,在培养物中的真核细胞中的表达也是本领域技术人员可用的,参见最近的综述,例如ref,(1993)curr.opinion biotech.4:573-576;trill et al.(1995)curr.opinion biotech 6:553-560。肽在酵母(例如,毕赤酵母)中的表达也是本领域众所周知的(参见,例如美国专利公开号20180142038、20190241645和20190119692)。
61、可以选择或构建合适的载体,该载体含有合适的调控序列,包括启动子序列、终止子序列、聚腺苷酸化序列、增强子序列、标志基因和视情况而定的其他序列。视情况而定,载体可以是质粒、病毒,例如噬菌体或噬菌粒。进一步的细节参见,例如,molecular cloning:a laboratory manual:2nd edition,sambrook et al.,1989,cold springharborlaboratory press。许多已知的核酸操作技术和方案,例如核酸构建体的制备、诱变、测序、将dna引入细胞和基因表达,以及蛋白质分析,在current protocols in molecularbiology,second edition,ausubel et al.eds.,john wiley&sons,1992或其随后的更新中有详细描述。
62、术语“宿主细胞”用于指代其中已经引入或能够引入编码本文所述的免疫调节多肽中的一种或多种的核酸序列并且进一步表达或能够表达选定的目的基因,诸如编码任何本文所述的纤溶酶抑制多肽的基因的细胞。该术语包括亲本细胞的后代,无论后代在形态学或遗传构成上是否与原始亲本相同,只要存在所选基因即可。因此,还考虑了包括将此类核酸引入宿主细胞的方法。引入可以采用任何可用的技术。对于真核细胞,合适的技术可以包括磷酸钙转染、deae-葡聚糖、电穿孔、脂质体介导的转染和使用逆转录病毒或其他病毒,例如痘苗病毒或杆状病毒(用于昆虫细胞)的转导。对于细菌细胞,合适的技术可包括氯化钙转化、电穿孔和使用噬菌体的转染。引入可以之后是引起或允许核酸表达,例如通过在用于表达基因的条件下培养宿主细胞。在一个实施方案中,核酸被整合到宿主细胞的基因组(例如染色体)中。根据标准技术,可以通过包含促进与基因组重组的序列来促进整合。
63、在某些实施方案中,本发明还提供了方法,其包括在表达系统中使用如上所述的构建体以表达特定多肽,诸如本文所述的纤溶酶抑制多肽。术语“转导”用于指基因从一种细菌转移到另一种细菌,通常通过噬菌体进行。“转导”也指通过逆转录病毒获取和转移真核细胞序列。术语“转染”用于指细胞摄取外来或外源性dna,并且当外源性dna已被引入细胞膜内时,细胞已被“转染”。许多转染技术是本领域众所周知的并且在本文中公开。参见,例如,graham et al.,1973,virology 52:456;sambrook et al.,2001,molecularcloning,a laboratory manual,cold spring harbor laboratories;davis et al.,1986,basic methods in molecular biology,elsevier;和chu et al.,1981,gene 13:197。此类技术可用于将一种或多种外源性dna部分引入合适的宿主细胞中。
64、如本文所用,术语“转化”是指细胞遗传特征的改变,并且当细胞被修饰以包含新的dna时,它已被转化。例如,细胞在从其天然状态进行基因修饰的地方被转化。在转染或转导之后,转化dna可以通过物理整合到细胞的染色体中而与细胞的dna重组,或者可以作为附加型元件暂时维持而不被复制,或者可以作为质粒独立复制。当dna随着细胞分裂而复制时,细胞被认为已经稳定转化。当与生物材料诸如核酸分子、多肽、宿主细胞结合使用时,术语“天然存在”或“天然的”是指在自然界中发现的且未被人操纵的材料。类似地,如本文所用,“非天然存在”或“非天然的”是指在自然界中未发现或已被人结构上修饰或合成的材料。
65、在某些实施方案中,本发明还涉及含有本文公开的纤溶酶抑制多肽的药物组合物。在一个实施方案中,药物组合物包含在药学上可接受的赋形剂、载体或稀释剂中的纤溶酶抑制多肽,并且当施用于动物,优选哺乳动物,最优选人时,其量有效抑制纤溶酶的至少一种活性。在其他实施方案中,药物组合物包含在药学上可接受的赋形剂、载体或稀释剂中的纤溶酶抑制多肽,并且其量有效治疗需要抑制纤溶酶活性的受试者,例如,在包括向受试者施用有效量的本文公开的纤溶酶抑制多肽的方法中。
66、与需要抑制纤溶酶有关的疾病、病症和治疗的实例包括但不限于外科手术、外伤,诸如创伤性脑损伤,以及其他病况和情况,诸如肿瘤发生、血管发生、骨重塑、血友病、冠状动脉旁路移植术(cabg)等。还考虑了包含本文所述的纤溶酶抑制多肽的药物组合物在其他情况下控制出血的用途,例如,作为抗纤维蛋白溶解组合物和作为纤溶酶过量或tpa或可直接或间接促进纤溶酶或其他相关蛋白酶的活性的其他血液学活性物质过量的解毒剂。
67、以纯形式或以合适的药物组合物的形式施用纤溶酶抑制多肽,可以通过任何可接受的用于类似用途的试剂的施用模式来进行。药物组合物可以通过将纤溶酶抑制多肽与合适的药学上可接受的载剂、稀释剂或赋形剂组合制备,并且可以配制成固体、半固体、液体或气体形式的制剂,诸如片剂、胶囊剂、散剂、颗粒剂、软膏剂、溶液剂、栓剂、注射剂、吸入剂、凝胶剂、微球剂和气雾剂。施用此类药物组合物的典型途径包括但不限于口服、表面、透皮、吸入、胃肠外、舌下、直肠、阴道、鼻内、腹膜内、静脉内、动脉内、透皮、舌下、皮下、肌内、直肠、经颊、鼻内、脂质体、经由吸入、眼内、经由导管(例如,在血管成形术中)、经由局部递送、皮下、脂肪内、关节内或鞘内。如本文所用,术语胃肠外包括皮下注射、静脉内、肌肉内、胸骨内注射或输注技术。配制药物组合物以允许其中含有的活性成分在将组合物施用于患者时是生物可利用的。将施用于受试者或患者的组合物采用一个或多个剂量单位的形式,其中例如,片剂可以是单一剂量单位,并且气雾剂形式的本发明化合物的容器可以容纳多个剂量单位。制备此类剂型的实际方法对本领域技术人员而言是已知的或显而易见的;例如,参见the scien20th edition(philadelphia college ofpharmacy and science,2000)ce and practice ofpharmacy,2000)。在任何情况下,要施用的组合物将含有用于根据本教导的治疗感兴趣的疾病或病况的治疗有效量的纤溶酶抑制多肽。
68、可用于本文的药物组合物还含有药学上可接受的载体,包括任何合适的稀释剂或赋形剂,其包括本身不诱导产生对接受组合物的个体有害的抗体的任何药剂,并且可以在没有过度毒性的情况下施用。药学上可接受的载体包括但不限于液体,如水、盐水、甘油和乙醇等。remington′s pharmaceutical sciences(mack pub.co.,n.j.current edition)中对药学上可接受的载体、稀释剂和其他赋形剂进行了全面讨论。
69、药物组合物可以是液体形式,例如酏剂、糖浆、溶液、乳剂或混悬剂。作为两个实例,液体可以用于口服施用或用于注射递送。当打算用于口服施用时,优选的组合物除本发明化合物外还含有一种或多种甜味剂、防腐剂、染料/着色剂和增味剂。在旨在通过注射施用的组合物中,可以包括一种或多种表面活性剂、防腐剂、润湿剂、分散剂、助悬剂、缓冲液、稳定剂和等渗剂。
70、液体药物组合物,无论是溶液、混悬液或其他形式,可以包括以下中的一种或多种辅料:无菌稀释剂,如注射用水、盐水溶液,优选生理盐水、林格氏溶液、等渗氯化钠、不挥发油,如可用作溶剂或悬浮介质的合成甘油单酯或甘油二酯、聚乙二醇、甘油、丙二醇或其他溶剂;抗菌剂,如苯甲醇或对羟基苯甲酸甲酯;抗氧化剂,如抗坏血酸或亚硫酸氢钠;螯合剂,如乙二胺四乙酸;缓冲液,如乙酸盐、柠檬酸盐或磷酸盐,以及用于调节张力的试剂,如氯化钠或葡萄糖。肠胃外制剂可以封装在由玻璃或塑料制成的安瓿瓶、一次性注射器或多剂量小瓶中。生理盐水是优选的佐剂。可注射药物组合物优选是无菌的。
71、用于胃肠外或其他施用的液体药物组合物应含有一定量的纤溶酶抑制多肽,以便获得合适的剂量。通常,该量是组合物中至少0.01%的纤溶酶抑制多肽。该量可以在组合物重量的0.1%至约70%之间变化。根据本发明的某些示例性药物组合物和制剂被制备成一个胃肠外剂量单位含有以重量计0.01%至10%的纤溶酶抑制多肽。
72、纤溶酶抑制多肽以治疗有效量施用,其将根据包括以下在内的多种因素而变化:特定多肽的活性纤溶酶抑制多肽的代谢稳定性和作用时间;患者的年龄、体重、一般健康状况、性别和饮食;施用方式和时间;排泄率;药物组合;特定疾病或病况的严重程度;和受试者正在接受的疗法。通常,治疗有效的每日剂量是(对于70kg哺乳动物)约1mg/kg(即70mg)至约10mg/kg(即7.0g);优选地,治疗有效剂量是(对于70kg哺乳动物)约2mg/kg至约8mg/kg;更优选治疗有效剂量为约4mg/kg。
73、本文提供的有效剂量范围不旨在限制并代表优选的剂量范围。然而,如相关领域技术人员所理解和确定的那样,最优选的剂量将针对个体受试者进行调整。(参见,例如,berkowet al.,eds.,the merck manual,16thedition,merck and co.,rahway,n.j.,1992;goodman et al.,eds.,goodman and gilman′s the pharmacological basis oftherapeutics,10thedition,pergamon press,inc.,elmsford,n.y.,(2001);avery′s drugtreatment:principles and practice of clinical pharmacology and therapeutics,3rd edition,adis press,ltd.,williams and wilkins,baltimore,md.(1987),ebadi,pharmacology,little,brown and co.,boston,(1985);osolci al.,eds.,remington′spharmaceutical sciences,18thedition,mack publishing co.,easton,pa.(1990);katzung,basic and clinical pharmacology,appleton and lange,norwalk,conn.(1992))。
74、如果需要,每次治疗所需的总剂量可以在一天、一周或一个月的过程中通过多剂量或单剂量施用。通常,治疗以小于纤溶酶抑制多肽的最佳剂量的较小剂量开始。此后,剂量以小的增量增加,直到达到环境下的最佳效果。纤溶酶抑制多肽可以单独施用或与针对病理学或针对病理学的其他症状的其他诊断剂和/或药物联合施用。施用纤溶酶抑制多肽的接受者可以是任何脊椎动物,如哺乳动物。在哺乳动物中,优选的受体是灵长目(包括人、猿和猴)、偶蹄目(包括马、山羊、牛、绵羊、猪)、啮齿目(包括小鼠、大鼠、兔和仓鼠)和食肉目(包括猫和狗)的哺乳动物。在鸟类中,首选的接受者是火鸡、鸡和同目其他成员。最优选的接受者是人。
75、可以将药物组合物配制成置于基质中,如可以置于体内的贴剂,使得纤溶酶抑制多肽随后从基质/贴剂释放并进入体内环境。此类的组合物可以包括例如背衬、活性化合物储库、控制膜、衬里和接触粘合剂。透皮贴剂可用于根据需要提供本发明纤溶酶抑制多肽的连续脉冲或按需递送。可适用于与本发明此类实施方案一起使用的示例性贴剂材料和方法在例如美国专利公开号20020049471、20110071498和20210038758中公开。
76、可以配制纤溶酶抑制多肽使得在通过使用本领域已知的程序对患者施用后提供活性成分的快速、持续或延迟释放。控释药物递送系统包括渗透泵系统和含有聚合物涂层储库或药物-聚合物基质制剂的溶解系统。受控释放系统的实例在美国专利号3,845,770和4,326,525以及p.j.kuzma et al.,regional anesthesia 22(6):543-551(1997)中给出,所有这些都通过引用并入本文。
77、最合适的途径将取决于所治疗病况的性质和严重程度。本领域技术人员还熟悉确定施用方法(例如口服、静脉内、吸入、皮下、直肠等)、剂型、合适的药物赋形剂和与将纤溶酶抑制多肽递送至需要抑制纤溶酶活性的受试者的其它事项。
78、根据各种预期的实施方案,需要抑制纤溶酶活性的受试者可以患有癌症(例如实体瘤,如肺癌、乳腺癌、前列腺癌或结肠癌,或另一种癌症)、血友病、类风湿性关节炎或全身炎症反应综合征(sirs)或被怀疑处于患的风险中,或者受试者可以需要或可以已经经历了血管生成、骨重塑或冠状动脉旁路移植术(cabg)。受试者可以正在接受手术或可以最近(例如,在1、2、4、6、8、10、12或24小时内,或在1、2、3、4、5、6、7、8、9或10天内)接受过手术,例如心血管手术、肿瘤手术、泌尿生殖外科手术、骨科手术、胸外科手术、整形外科手术、创伤手术、腹部手术、移植手术、神经外科手术或耳鼻喉科手术。
79、相关领域的技术人员将熟悉任何数量的本文所述的药物组合物的施用可以适应其的诊断、手术和其他临床标准。参见,例如,humar et al.,atlas of organtransplantation,2006,springer;kuo et al.,comprehensive atlas oftransplantation,2004lippincott,williams&wilkins;gruessner et al.,living donororgan transplantation,2007 mcgraw-hill professional;antin et al.,manual ofstem cell and bone marrow transplantation,2009 cambridge university press;wingard et a1.(ed.),hematopoietiic stem cell transplantation:a handbook forclinicians,2009 american association of blood banks;sabiston,textbook ofsurgery,2012 saunders&co.;mulholland,greenfield′ssurgery,2010 lippincott,williams&wilkins;schwartz′s principles of surgery,2009 mcgraw-hill;lawrence,essentials of general surgery 2012 lippincott,williams&wilkins。
80、上文中的参考文献
81、(1)fergusson da,hebert pa,mazer cd,fremes s,macadams c,murkin jm,etal.a comparison of aprotinin and lysine analogues in high-risk cardiacsurgery.n engl j med 2008;358:2319-2331.
82、(2)karkouti k,wij eysundera dn,yau tm,mccluskey sa,tait g,beattiews.the risk-benefit profile of aprotinin versus tranexamic acid in cardiacsurgery.anesth analg 2010;110:21-9.
83、(3)martin k,knorr j,breuer t,gertler r,macguill m,lange r,tassani p,wiesner g.seizures after open heart surgery:comparison of ε-aminocaproic acidandtranexamic acid.j cardiothorac vasc anesth.2011;25:20-25.
84、(4)lecker i,wang ds,whissell pd,avramescu s,mazer cd,orserba.tranexamic acid-associated seizures:causes and treatment.ann neurol.2016;79:18-26.
85、(5)stevens lm,noiseux n,prieto i,hardy jf.major transfusions remainfrequent despite the generalized use of tranexamic acid:an audit of 3322pattients undergoing cardiac surgery.transfusion 2016;56:1857-65.
86、(6)faraoni d,levy jh.development of a novel blood-sparing agent incardiac surgery:do we need another agent?annesth ahalg.2014;119:11-12.
87、(7)bokesch pm,szabo g,wojdyga r,grocott hp,smith pk,mazer cd,vetticaden s,wheeler a,levy jh.a phase 2prospective,randomized,double-blindtrial comparing the efiects of tranexamic acid with ecallantide on bloodlossfrom high-risk cardiac surgery with cardiopulmonary bypass(conserv-2trial).j thorac cardiovasc surg.2012;143:1022-1029.
88、(8)englberger l,dietrich w,eberle b,erdoes g,keller d,carrel t.anovel blood-sparing agent in cardiac surgery?first in-patient experience withthe synthetic serine protease inhibitor mdco-2010:a phase ii,randomized,double-blind,placebo-c0ntrolled study in patients undergoing coronary arterybypass grafting with cardiopulmonarybypass.anesth analg.2014;119:16-25.
89、(9)earl s t,masci pp,de jersey j,lavin mf,dixon j.drug developmentfrom australian elapid snake venoms and the venomics pipeline ofcandidatesforhaemostasis:textilinin-1(q8008),haempatchtm(q8009)andcovasetm(v0801).toxicon.2012;59:456-463.
90、(10)beith jg.in vivo significance of kinetic constants of proteinproteinase inhibitors.biochem med 1984;32:387-97.
91、(11)morrison jf,walsh ct.the behavior and significance of slow-binding enzyme inhibitors.adv enzymol relat areas mol biol 1988;61:201-301.
92、(12)kawamura k,yamada t,kurihara k,tamada t,kuroki r,tanaka i,takahashi h,niimura n.x-ray and neutron protein crystallographic analysis ofthe trypsin-bpti complex.acta crystallogr d biol crystallogr.2011;67:140-148.
93、(13)fujikawa k,chung dw,hendrickson le,davie ew.amino acid sequenceof human factor xi,a blood coagulationfactor with four tandem repeats thatare highly homologous with plasma prekallikrein.biochemistry.1986;25:2417-2424.
94、(14)leytus sp,foster dc,kurachi k,davie ew.gene for human factor x:ablood coagulation factor whose gene organization is essentially identicalwith that of factor ix and protein c.biochemistry.1986;25:5098-5102.
95、实施例1:2型人组织因子途径抑制剂结构城1的纤溶酶特异性的kunitz抑制剂(具有c端iek的60个残基y11t/l17r双突变体)的增强的抗纤维蛋白溶解功效
96、当前的抗纤维蛋白溶解剂通过抑制纤溶酶活性位点(例如,抑肽酶)或通过防止纤溶酶原/组织纤溶酶原激活剂(tpa)与纤维蛋白凝块结合来减少失血(例如,ε-氨基己酸和氨甲环酸);然而,它们有不良副作用。在这里,我们表达了2型人组织因子途径抑制剂的60个残基kunitz结构域1(kd1)突变体,该突变体能够抑制纤溶酶以及纤溶酶原激活。单突变体(kd1-l17r-kt)和双突变体(kd1-y11t/l17r-kt)在大肠杆菌中表达为his标记的构建体,每个构建体都有肠激酶切割位点。还在巴斯德毕赤酵母中表达kd1-y11t/l17r-kt。kd1-y11t/l17r-kt抑制纤溶酶的作用与抑肽酶相当,并结合纤溶酶原/纤溶酶和tpa的kringle结构域,kd分别为~50nm和~35nm。重要的是,与抑肽酶相比,kd1-l17r-kt和kd1-y1 1t/l17r-kt不抑制激肽释放酶。此外,血浆凝块溶解测定中,kd1-y11t/l17r-kt的抗纤维蛋白溶解能力优于kd1-l17r-kt,与抑肽酶相似。在血栓弹性成像实验中,kd1-y11t/l17r-kt以剂量依赖性方式抑制纤维蛋白溶解,并且与较高浓度的抑肽酶相当。此外,kd1-y11t/l17r-kt在原代人内皮细胞或成纤维细胞中不诱导细胞毒性。我们得出结论,kd1-y11t/l17r-kt与已知最有效力的纤溶酶抑制剂抑肽酶相当,并且能使用毕赤酵母来大量生产。
97、1.简介
98、在严重创伤中和重大手术过程(如心脏外科手术)中,纤维蛋白溶解系统过度激活,导致大出血(1-3)。在战场、事故和医院环境中,大出血会带来重大的死亡风险和成本。不受控制的出血是创伤中可预防死亡的主要原因,并且经常导致外科手术期间需要大量输血(4,5)。抗纤维蛋白溶解剂通过抑制纤局维蛋白溶解,从而抑制纤维蛋白降解产物,减少输血需求(6,7)。抑肽酶(牛胰胰蛋白酶抑制剂,bpti)是纤溶酶活性位点的有效的抑制剂,一直是减少心脏外科手术和肢体创伤期间失血的主要抗纤维蛋白溶解剂(8)。然而,它的使用与严重的副作用有关,如肾损伤、心肌梗塞和中风(9-10)。此外,抑肽酶来源于牛,其潜在的过敏反应是一个主要问题(11)。由于这些原因,它于2008年暂时退出临床市场(12)。目前批准的治疗剂氨甲环酸(txa)和ε-氨基己酸(eaca)是赖氨酸类似物,可避免纤溶酶原和组织纤溶酶原激活剂(tpa)与纤维蛋白凝块结合(13,14)。结果,部的纤溶酶原向纤溶酶的激活被禁止,纤维蛋白溶解被阻止。然而,eaca和txa在减少失血方面不如抑肽酶有效(15)。此外,与抑肽酶一样,它们也会导致肾功能衰竭(16),最近的证据表明txa和程度较轻的eaca与癫痫发作的显著发生率相关(16,17)。因此,需要没有抑肽酶和赖氨酸类似物的副作用的改进的抗纤维蛋白溶解剂。
99、文献中报道了几种活性位点纤溶酶抑制剂(18-25),但其中大部分的开发阶段未知。textilinin-1(q8008),来自东部拟眼镜蛇(pseudonajja textilis)的kunitz结构域纤溶酶活性位点抑制剂(19,26),正处于临床前开发阶段。q8008抑制纤溶酶的亲和力比抑肽酶弱10到15倍,但它对激肽释放酶的抑制也很弱(19,26)。在小鼠尾部出血模型中,据报道q8008在减少失血方面与抑肽酶一样有效(26)。然而,由于q8008来源于蛇毒,它可以在人中引起类似于对于抑肽酶观察到的过敏反应。此外,使用向日葵胰蛋白酶抑制剂-1的支架,设计了非常有效力的纤溶酶的环肽活性位点抑制剂,其ki为0.05nm,并已被提议作为药物开发的候选(25)。此外,还有变构合成纤维蛋白溶解抑制剂被提议用于减少围手术期出血,但它们处于非常早期的开发阶段(27,28)。此外,非常有效力的纤溶酶抑制剂(dx-1000)正在被开发为抗肿瘤剂而不是抗纤维蛋白溶解剂(29)。
100、值得注意的是,当抑肽酶在2008年被禁止时,对抑制激肽释放酶和纤溶酶的药理剂艾卡拉肽(ecallantide)(dx-88)进行了临床评估(30)。由于在ecallantide组中观察到死亡率增加,该研究提前终止。另一种抑制纤溶酶、因子(f)xa、fxia和激活的蛋白c(apc)的试剂mdco2010也进行了临床评估(31)。由于治疗组中严重不良事件的数量增加,该研究也提前终止。安全问题的原因和与药物使用的潜在联系正在进一步调查中。
101、在本实施例中,我们描述了抗纤维蛋白溶解剂,它以与抑肽酶相当的效力抑制纤溶酶,但它是非常弱的激肽释放酶抑制剂。它是使用2型人组织因子途径抑制剂(tfpi-2)的kunitz结构域1(kd1)作为支架设计的。新的60个残基纤溶酶抑制剂kd1-y11t/l17r-kt与早期的具有c端iekvpk的异质单突变体kd1-l17r(标示为kd1-l17r-kcooh)相比具有一个额外的突变具有一个不同的c端赖氨酸(iekt)(32)。kd1-y11t/l17r-kt比目前的单突变体kd1-l17r-kt更好地抑制纤溶酶,除纤溶酶外,它还结合纤溶酶原的kringle结构域和tpa,解离常数为35至50nm。在血浆中2μm的治疗剂量,抑肽酶的最低hammersmith方案(33)下,因此预计kd1-y11t/l17r-kt通过抑制纤溶酶活性位点以及阻断纤溶酶原和tpa与纤维蛋白凝块结合来有效抑制纤维蛋白溶解。因此,kd1-y11t/l17r-kt看起来是在临床环境中替代抑肽酶的有前途的候选。本文提供了比较抑肽酶与kd1-y11t/l17r-kt的实验细节。此外,建模用于评估tyr11至thr的突变以及在tfpi-2kd1抑制剂支架中的c端的iek的影响。讨论了从此类建模中获得的结构信息,以描述突变体的增强的抗纤维蛋白溶解活性。
102、2.实验部分
103、2.1.材料
104、大肠杆菌(e.coli)菌株bl21(de3)plyss和pet28a表达载体获自novagen inc.(madison,wi)。amicon离心过滤装置(3000mr截断值)购自millipore(bedford,ma)。qsepharose ff、superdex 200和his-trap hp柱获自amersham biosciences。二异丙基氟磷酸盐(dfp)来自calbiochem(san diego,ca)。txa、eaca、卡那霉素和异丙基硫代半乳糖苷(iptg)购自sigma(st.louis,mo)。半胱天冬酶-glo 3/7测定试剂盒和celltoxtm green细胞毒性测定试剂盒来自promega(madison,wi)。纯化的人fxia、凝血酶(iia)和纤溶酶购自hematologic technologies inc(essex junction,vt)。血浆激肽释放酶(pklk)来自酶研究实验室(south bend,in)。阿替普酶(alteplase)(tpa)购自genentech(south sanfrancisco,ca)。重组肠激酶来自novogen,emd chemicals(san diego ca)。正常混合血浆(npp)购自george king bio-medical inc.(overland park,kansas)。δ纤溶酶(含有蛋白酶结构域和第一个kringle结构域的重组纤溶酶)获自victor marder博士(university ofcalifomia,los angeles,ca),紫杉醇由zhenfeng duan博士(university of califomia,los angeles,ca)提供。抑肽酶(bpti)购自zymogenetics(seattle,wa),人因子viia(fviia)如先前所描述的制备(34)。可溶性组织因子(stf,残基1-219)获自tom girard(washington university,st.louis,missouri)。纤溶酶底物s-2251(h-d-val-leu-lys-对硝基苯胺)、pklk和fxia底物s-2366(pyroglu-pro-arg-对硝基苯胺)和fviia底物s-2288(h-dile-pro-arg-对硝基苯胺)购自diapharma inc(west chester,oh)。新鲜的正常人柠檬酸化血液来自nebraska medical center,omaha。每个献血者的部分促凝血酶原激酶时间(pt)和激活的部分促凝血酶原激酶时间(aptt)均正常。
105、2.2.kd1-l17r-kt和kd1-y11t/l17r-kt在大肠杆菌中的表达和纯化
106、克隆了含有c端iek的kd1-l17r-kt和kd1-y11t/l17r-kt的cdna序列,并使用t7启动子系统在大肠杆菌菌株bl21(de3)plyss中作为氨基端his6标记的融合蛋白过表达。根据标准程序制备衍生自pet28a的重组质粒,该质粒包含his6前导序列,随后是肠激酶切割位点和编码kd1-l17r-kr或kd1-y11t/l17r-kt的cdna(35)。表达的构建体的序列在图10中给出。his6标记的kd1-l17r-kt和kd1-y11t/l17r-kt在大肠杆菌中表达,所述大肠杆菌在含有15mg/升的卡那霉素的luria肉汤中生长,并在37℃下用1mm iptg在对数中期(a600~0.9)诱导5-6小时。his6标记的kd1-l17r-kt和kd1-y1 1t/l17r-kt使用充镍的his-trap柱从包涵体纯化。his-trap纯化的蛋白质使用还原和氧化的谷胱甘肽系统重新折叠,并使用q-sepharose ff柱进一步纯化,如先前所记载的(32,36)。
107、2.3.在巴斯德毕赤酵母中kd1-y11t/l17r-kt克隆构建及表达
108、巴斯德毕赤酵母(pichia pastoris)菌株x-33和分泌性表达载体ppiczαa购自invitrogen(san diego,ca)。对应于氨基酸序列(图10)的kd1-y11t/l17r-kt cdna由idt(coralville,ia)合成。通过pcr扩增cdna并将产物线性化,并亚克隆到ppiczαa的xhoi和noti限制性位点。在dh5α感受态细胞中进行进一步的载体扩增。使用bio-rad gene pulser电穿孔仪经由电穿孔将提取的cdna引入巴斯德毕赤酵母x-33。将转化子铺板在补充有500μg博来霉素(zeocin)/ml的ypd板上。通过sds-page评估菌落在bmm培养基中的kd1-y11t/l17r-kt表达。使用缓冲的基本甘油培养基(bmgy)ph 6.0制备发酵接种摇瓶。首先,将表达kd1-y11t/l17r-kt的单个菌落接种到50ml中持续12小时,然后将0.5ml所得培养物转移到300ml bmgy ph 6.0中持续20小时。然后将后者接种到含有3l基础盐培养基(bsm)ph 5.0的15l nlf bioengineering生物反应器(wald,switzerland)中。用甲醇诱导蛋白质表达,并在30℃、ph 5.0和40%溶解氧下进行48小时。发酵肉汤在4℃下以7200rpm离心。收集含有kd1-y11t/l17r-kt的上清液并储存在-30℃直至进一步处理。
109、2.4.从巴斯德毕赤酵母中纯化kd1-y11t/l17r-kt
110、将100ml发酵上清液调至ph 8.5并在室温下以1500rfc离心5分钟。然后,收集上清液并调节至ph值3.0,并与0.5ml tritonx-100混合30分钟。添加尿素高至终浓度为4m,并在室温下孵育2.5小时。孵育后,将溶液稀释至最终电导率为12ms。使用biocad vision工作站以120cm/小时的恒定流速进行纯化。将样品加载到预先用50mm磷酸盐缓冲液ph 2.8(洗涤缓冲液)平衡过的sp-sepharose(ge healthcare bio-sciences pittsburgh,pa)柱上。然后,用两倍柱体积的洗涤缓冲液洗涤柱,并用含有1.0m nacl的50mm磷酸盐缓冲液(ph2.8)洗脱蛋白质。合并含有kd1-y11t/l17r-kt的级分,并补充l-精氨酸至0.5m和甘露醇至7%。在室温孵育1小时后,使用3.5kda mw截断值膜透析管(spectrum.nj)针对10mm磷酸盐缓冲液ph 8.0进行透析。
111、2.5.sds-page
112、使用laemmle缓冲系统(37)进行sds-page。使用的丙烯酰胺浓度为15%,凝胶用考马斯亮蓝染料染色。
113、2.6.蛋白酶抑制测定
114、所有反应均在tbs,ph 7.5(50mm tris-hcl,ph 7.5,含有100mm nacl)中进行,其中含有0.1mg牛血清白蛋白/ml(tbs/bsa)和2mm ca2+(tbs/bsa/ca2+,ph 7.5)。每种酶(纤溶酶、pklk、fxia或fviia/stf)与不同浓度(10-1至2x 103nm)的kd1-wt、kd1-l17r-kt、kd1-y11t/l17r-kt或抑肽酶(bpti)一起在96孔微量滴定板(总体积100μl/孔)中室温下孵育1小时。然后将适合每种酶的合成底物(5μl)添加至1km的最终浓度,并在vmax动力学酶标仪(molecular devices)中测量残留的酰胺分解活性。使用非线性回归数据分析程序grafit确定抑制常数ki*。抑肽酶、kd1-wt、kd1-l17r-kt和kd1-y11t/l17r-kt的数据使用紧密结合抑制剂的等式(等式1)进行分析,其中vi和v0分别是抑制率和未抑制率,(i)0和(e)0分别是抑制剂和酶的总浓度(38,39)。
115、
116、根据beith(38),使用等式2,通过校正底物的影响获得ki值,其中(s)是底物浓度,km对每种酶是特异的。
117、
118、2.7.dip-δ纤溶酶的制备
119、通过在室温下用等体积的1m tris-hcl、ph 8.0和1m dfp(最终浓度为1mm dfp)处理δ纤溶酶20分钟,然后在冰上孵育几小时来产生活性位点阻断的δ纤溶酶。加入额外等体积的1m tris-hcl,ph 8.0和1m dfp(最终浓度为2mm)并将反应物在室温下孵育20分钟,然后在4℃下过夜。dfp抑制的δ纤溶酶(dip-δ纤溶酶)针对含有150mm nacl的20mm hepes ph7.5进行透析,并使用s-2251合成底物水解测定其残留活性。基于残留活性,>99%的δ纤溶酶失活。当使用sds-page分析时,dip-δ纤溶酶显示没有蛋白质降解。
120、2.8.使用表面等离子体共振(spr)kd1-y11t/l17r-kt与tpa和dip-δ纤溶酶的结合
121、结合研究在biacore t100流量生物传感器(biacore,uppsala,sweden)上在25℃下进行。使用胺偶联化学将dip-δ纤溶酶(使用sds-page的纯度约为98%)或tpa(使用sds-page的纯度>98%)固定在羧甲基-右旋糖酐流动池(cm5传感器芯片,ge healthcare)上。用1-乙基-3-(3-二甲基氨基丙基)碳二亚胺和n-羟基磺基琥珀酰亚胺的混合物活化流动池表面5分钟(流速10μl/分钟),然后将蛋白质(20μg/ml于10mm乙酸钠,ph 5.5中)注射到表面上。未反应的位点用1m乙醇胺封闭5分钟。hbs-p缓冲液(20mm hepes,ph 7.4、100mm nacl、0.005%(v/v)p20)中的分析物kd1-y11t/l17r-kt(100至2000nm)以10μl/分钟灌流通过流动池持续6分钟。在更换为不含蛋白质的hbs-p缓冲液后,监测分析物解离持续10分钟。用含有20mm eaca的hbs-p再生流动池。通过减去灌流通过无偶联蛋白的流动池的分析物获得的信号针对非特异性结合校正数据。使用biaevaluation软件(biacore)使用1:1结合模型分析结合。kd值是由导出的解离(kd)和缔合(ka)速率常数的商计算的。
122、2.9.纤维蛋白溶解(凝块溶解)测定
123、使用sperzel和huetter(40)的方法,如前所述(32,41)稍作修改。简而言之,iia用于启动npp中的纤维蛋白形成,并通过同时添加tpa诱导形成的凝块的溶解(纤维蛋白溶解)。用moleculardevices酶标仪(spectramax 190)测量405nm处的光密度来监测凝块形成和溶解。简而言之,将10μl每种测试化合物(kd1-l17r-kt、kd1-y11t/l17r-kt、抑肽酶)或盐水对照添加到240μl npp中。然后将225μl的这种混合物添加到含有25mm cacl2的tbs/bsa中的25μl iia和tpa。在250μl的最终体积中,iia的浓度为0.15μg/ml,tpa的浓度为1μg/ml。在对照条件下(零tpa和零测试化合物),od405立即增加表明凝血,随后极其缓慢的下降表明纤维蛋白溶解。由于凝血在5分钟后几乎完成,tpa诱导的纤维蛋白溶解被评估为长达60分钟的od405的相对降低。kd1-l17r-kt在0.5μm至5μm的最终浓度下进行了测试,而kd1-y11t/l17r-kt和抑肽酶在0.5μm至3μm的最终浓度下进行了测试。
124、2.10.血栓弹性成像
125、使用teg 5000 thrombelastograph(haemonetics corp,braintree,ma)通过血栓弹性成像(teg)评估了不同浓度的kd1-wt、kd1-l17r-kt、kd1-y11t/l17r-kt、抑肽酶或eaca对纤维蛋白溶解的影响。每个凝块形成/溶解测定包含300μl柠檬酸化全血;纤溶酶(1.5μm最终浓度)、cacl2(10mm最终浓度)和各种浓度的林格溶液中的每种抗纤维蛋白溶解剂,使最终体积达到360μl。最后添加纤溶酶和cacl2以同时启动凝血和纤维蛋白溶解。基于纤溶酶对凝块强度和溶解的影响,选择1.5μm纤溶酶浓度。每个实验进行180分钟以确定ly60值。每天校准血栓弹性成像,并一式两份地测试每种抑制剂浓度。teg分析软件(4.2.2版;haemonetics corporation,braintree,ma)用于计算凝块起始时间(r)、最大凝块强度(最大幅度(ma),这与剪切弹性模量强度,g直接相关),以及ma后60分钟的溶解百分比(ly60)(42)。
126、2.11.细胞毒性测定
127、2.11.1.细胞和培养条件
128、原代人混合脐静脉内皮细胞(huvec)获自atcc。将细胞维持在补充有内皮细胞生长试剂盒-bbe(atcc)和青霉素-链霉素-两性霉素b(atcc)的血管细胞基础培养基(atcc)中。原代人皮肤成纤维细胞获自lonza,并维持在补充有生长因子、胎牛血清和抗生素的混合物(fgmtm-2singlequotstm,lonza)的成纤维细胞基础培养基(fbmtm,lonza)中。所有细胞都保持在37℃的湿润5%co2气氛中,并在达到80%汇合后传代。所有实验均使用处于对数生长期的细胞进行。
129、2.11.2.抗纤维蛋白溶解剂(kd1-y11t/l17r-kt、抑肽酶、eaca和txa)
130、在磷酸盐缓冲液中制备抗纤维蛋白溶解剂的储备溶液。对于毒性研究,将细胞以3,500个细胞/cm2接种到96孔或24孔细胞培养板中,并在它们达到80%汇合后用于实验。用以下浓度的抗纤维蛋白溶解剂处理细胞24小时——抑肽酶和kd1-y11t/l17r-kt为0.1μm、1μm、10μm和30μm;eaca为1mm、5mm、20mm和60mm和txa为0.2mm、2mm、10mm和30mm。
131、2.11.3.刃天青还原测定
132、刃天青还原测定(fisher scientific)用于评估抗纤维蛋白溶解剂对原代人内皮细胞和皮肤成纤维细胞的潜在细胞毒性。用于溶解样品的磷酸盐缓冲液作为阴性对照被包括在内。该测定基于由存活细胞将非荧光染料刃天青还原为高荧光的resorufin。荧光信号与活细胞的数量成比例,因为非存活细胞无法还原染料并且不会产生荧光信号。简而言之,96孔细胞培养板中的细胞用不同浓度的抗纤维蛋白溶解化合物处理(如上所述)。24小时后,将刃天青试剂添加到每个孔中,并将板在37℃下孵育4小时。使用544nm的激发波长和590nm的发射波长,通过fluostar omega酶标仪(bmg labtech)测量荧光。每个测定一式两份进行,每个重复三次。基于与未处理细胞的比较来评估生存力。
133、2.11.4.半胱天冬酶3/7测定
134、使用半胱天冬酶-glo 3/7测定试剂盒(promega)检测抗纤维蛋白溶解剂在细胞中对凋亡的影响。半胱天冬酶3和7在经历凋亡的细胞中被激活。该测定为半胱天冬酶3和7提供发光底物。酶活性导致发光,这与存在的半胱天冬酶活性量成比例。将细胞接种在96孔板中,并用抗纤维蛋白溶解剂或磷酸盐缓冲液(溶剂对照)处理。紫杉醇作为阳性对照被包括在内。处理24小时后,每孔加入半胱天冬酶试剂,混匀,室温孵育1小时。使用fluostaromega酶标仪(bmg labtech)测量发光。
135、2.11.5.细胞毒性测定
136、使用celltoxtm green cytotoxicity assay(promega)评估细胞毒性和细胞凋亡。该测定测量因细胞死亡而发生的膜完整性变化。系统中使用的染料从活细胞中排出,但与受损细胞中的dna结合,从而产生荧光信号。暴露24小时后,我们在24孔板中测量了用4个浓度(如上所示)的抗纤维蛋白溶解剂处理的huvec和原代成纤维细胞中的细胞凋亡,每个浓度一式三份。hoechst(thermo fisher scientific)用于染色所有细胞核。使用倒置显微镜(nicon;edipse t2000 te)捕获细胞图像。使用image j软件对绿色荧光细胞(fitc过滤器)和hoechst染色细胞(dapi过滤器)进行计数。荧光细胞显示为所有细胞的百分比。
137、2.12.统计方法
138、单因素方差分析(anova)用于比较血浆凝块溶解测定中抗纤维蛋白溶解剂(kd1-wt、kd1-l17r-kt、kd1-y11t/l17r-kt、抑肽酶)在抑制纤维蛋白溶解方面的作用。用于比较任何两个平均值的p值是使用事后检验计算的,并使用tukey调整针对多重比较进行了调整。对于teg数据,levene的f检验显示未满足方差同质性。因此,使用了welch的f检验并进行了games-howell事后程序以确定哪些对的平均ma和平均ly60%水平存在显著差异。对于细胞毒性测定,通过anova分析收集的数据集,并使用学生t检验比较各个组。所有实验重复两次或三次,结果相似。定量值报告为平均值±标准偏差(sd)或平均值的标准误差(sem),如图图例所示。在0.05或更低的p值下,差异被认为具有统计学意义。所有统计分析均使用spssv27(ibm corp.,armonk,ny,usa)进行。
139、2.13.分子建模
140、δ-纤溶酶(43)、纤溶酶原kringle结构域1(14)和野生型kd1(36)的晶体结构被用作模板来模拟kd1-y11t/l17r-kt与δ-纤溶酶和与纤溶酶kringle结构域1的复合物。先前已经描述了用于对这些复合物进行建模的方案(41,44)。由于野生型kd1晶体结构中的c端残基是无序的,我们使用modeller程序(45)来构建kd1-y11t/l17r-kt分子的这一部分。使用amber程序(46),在10的谐波约束下,对构建的模型进行1000步最小化,从而进一步细化构建的模型。
141、3.结果
142、3.1.kd1-l17r-kt和kd1-y11t/l17r-kt在大肠杆菌中的表达和纯化
143、60个残基的his6标记的kd1-l17r-kt和kd1-y11t/l17r-kt在具有肠激酶切割位点的大肠杆菌菌株bl21(de3)plyss中表达(图10)。与先前表达的在c端具有iekvpk的kd1-l17r(标示为kd1-l17r-kcooh)(41)相比,这些构建体在n端短了9个残基,在以iekt结尾的c端短了3个残基(图10)。使用q-sepharose ff柱重新折叠和纯化融合蛋白。纯化的kd1突变蛋白与肠激酶一起孵育以去除his6-标签;然而,在酶与底物的比例为1:50时,切割不成功。his6-标签去除不成功的原因可能是由于kd1突变体对肠激酶的抑制类似于抑肽酶对肠激酶的抑制(47)。图10示出了纯化的在nh2端均含有肠激酶切割位点和his6-标签的kd1-l17r-kt和kd1-y11t/l17r-kt的sds-page分析。
144、3.2.kd1-y11t/l17r-kt在巴斯德毕赤酵母中的表达和纯化
145、由于在大肠杆菌表达的突变体中,his6标签不能被肠激酶去除,我们使用巴斯德毕赤酵母表达了60个残基双突变体kd1-y11t/l17r-kt,并如实验部分所述纯化至同质。从100ml培养基中纯化出大约50mgkd1-y11t/l17r-kt。纯化的巴斯德毕赤酵母kd1-y11t/l17r-kt的sds-page分析如图11所示。请注意,与相应的大肠杆菌表达的含有his6标签和肠激酶溶解序列的kd1-y11t/l17r-kt相比,巴斯德毕赤酵母表达的kd1-y11t/l17r-kt的mw略低(图11)。
146、3.3.kd1-l17r-kt和kd1-y11t/l17r-kt的抑制概况
147、含有iia切割位点的野生型kd1(kd1-wt)以6.0±0.5nm的ki抑制纤溶酶(32,图12a)。具有iek c端的含有肠激酶切割位点的kd1-l17r-kt以0.9±0.1nm的ki抑制纤溶酶,这与之前描述的具有vpk c端的含有iia切割位点的kd1-l17r-kcooh相似(41)。大肠杆菌和巴斯德毕赤酵母表达的kd1-y11t/l17r-kt(ki0.59±0.1)二者均以与抑肽酶(ki0.49±0.1)相似的亲和力抑制纤溶酶(图12a)。表1中提供了每种抑制剂对纤溶酶抑制的ki值。因此,肠激酶切割序列和his6-标签不影响抑制活性。此外,类似于kd1-l17r-kcooh(32,41),kd1-y11t/l17r-kt(本实例)微弱地抑制fviia/stf、fxia和pklk,ki>3μm(图12b)。
148、表1.kd1-wt、kd1-y11t-kt、kd1-l17r/y11t-kt和抑肽酶抑制纤溶酶的ki值。
149、
150、*ki值表示三个独立测量的平均±sd
151、3.4.kd1-y11t/l17r-kt与dip-δ纤溶酶和tpa的结合。
152、我们使用了spr来研究kd1-y11t/l17r-kt与固定化dip-δ纤溶酶(图13a)和tpa(图13b)的结合。dip-δ纤溶酶与kd1-y11t/l17r-kt结合的kon为1.49±0.3x103m-1s-1;koff为7.13±0.9x10-5s-1,kd为47.6±7nm。tpa与kd1-y11t/l17r-kt结合的kon为2.91±0.4x103m-1s-1;koff为1.05±0.7x10-4s-1,kd为35.4±5nm。
153、3.5.纤维蛋白溶解(凝块溶解)测定
154、进行这些实验是为了比较kd1-l17r-kt、kd1-y11t/l17r-kt和抑肽酶在抑制tpa诱导的血浆凝块纤维蛋白溶解方面的有效性。向npp添加iia导致纤维蛋白形成,这通过od405的增加反映出来(曲线iia,零tpa,图14a-c)。同时添加tpa导致初始凝块形成,随后是由tpa介导的纤溶酶原转化为纤溶酶诱导的纤维蛋白溶解(曲线iia,tpa;图14a-c);在不存在纤维蛋白溶解抑制剂的情况下,每种情况下的纤维蛋白溶解中点都在6到7分钟之间。所有三种试剂都以剂量依赖的方式抑制纤维蛋白溶解。表2中提供了在所用抑制剂的每个浓度下的最大od405、60分钟时的od405和达到纤维蛋白溶解中点的时间。最大od405在抑制剂之间以及所用抑制剂的浓度之间没有差异。最大od405反映了iia诱导的形成的纤维蛋白凝块的强度,这在随后由tpa在凝块位点产生的纤溶酶开始溶解之前迅速实现。因此,预计使用的每种抑制剂在不同浓度下的最大od405将是相似的。进一步,60分钟时的od405表明纤维蛋白溶解的程度,这对于较低浓度的每种抑制剂来说相对相似;然而,与较高浓度的抑肽酶相比,kd1-l17r-kt的减少更多,kd1-y11t/l17r-kt的减少适度(图14,表2)。
155、重要的是,kd1-l17r-kt分别在0.5μm时将纤维蛋白溶解中点从约7分钟增加到约10分钟,在1μm时增加到约13分钟,在1.5μm时增加到约17分钟,在3μm时增加到约31分钟,在4μm时增加到约43分钟,并且在5μm时增加到约55分钟(图14a,表2)。kd1-y11t/l17r-kt分别在0.5μm时将纤维蛋白溶解中点从约7分钟增加到约12分钟,在1μm增加到约28分钟,在1.5μm时增加到约43分钟,在2μm和3μm时增到>60分钟(图14b,表2)。抑肽酶分别在0.5μm时将纤维蛋白溶解的中点从约7分钟增加到约13分钟,在1μm时增加到约40分钟,在1.5μm和>1.5μm浓度下增加到>60分钟(图14c,表2)。累积地,图15中显示的统计分析表明,与kd1-l17r-kt相比,kd1-y11t/l17r-kt在增加纤维蛋白溶解中点方面更有效,抑肽酶比kd1-y11t/l17r-kt稍微更有效。
156、3.6.血栓弹性成像
157、进行血栓弹性成像实验以评估kd1-wt(31)、kd1-l17r-kt、kd1-y11t/l17r-kt、抑肽酶和eaca对纤溶酶诱导的通过添加cacl2在全血中形成的凝块的溶解的影响。这些数据呈现在图16中并总结在表3中。图16a显示了不同浓度的纤溶酶对cacl2启动的凝块形成的teg迹线。在没有纤溶酶的情况下,达到的平均最大幅度(ma)约47mm,剪切弹性模量强度g约为4620dyn/cm2,并且在60分钟时未检测到凝块溶解(ly60<0.1%)。在1.5μm纤溶酶时,ma达到约7mm,g值约401dyn/cm2,并且30分钟内发生100%的凝块溶解(表3)。在>1.5μm纤溶酶时,未观察到凝块形成。图16b-f说明了kd1-wt、kd1-l17r-kt、kd1-y11t/l17r-kt和抑肽酶在不同浓度(1μm至7.5μm)下对凝块形成和在1.5μm纤溶酶存在下的溶解的平均teg迹线。数据表明,所有测试的抗纤维蛋白溶解剂均以浓度依赖性方式改善凝块硬度(ma)、剪切强度(g)并抑制纤维蛋白溶解(表3)。值得注意的是,在抑制剂浓度为5μm(对应于hammersmith方案的高剂量,这是已建立的抑肽酶临床施用方案)时(33),kd1-y11t/l17r-kt将凝块强度ma提高至约80%(37.5mm)和g提高至约65%(约3004dyn/cm2,而抑肽酶将ma提高至约69%(32.9mm)并将g提高至约53%(约2453dyn/cm2)。然而,kd1-y11t/l17r-kt检测到ly60约为12%,与抑肽酶为0.2%相比。在7.5μm浓度下,kd1-y11t/l17r-kt和抑肽酶二者具有相似的ma(约83%和约80%)和g(约70%和约65%),以及ly60(各自为0.2%)。eaca也以剂量依赖性方式提高了ma、g和ly60(图16g)。然而,在eaca浓度为3mm(在临床环境中使用的剂量)时,它仅将ma和g分别提高至约67%和约50%。总的来说,teg数据表明eaca在恢复ma和g方面不如kd1-y11t/l17r-kt或抑肽酶有效。进一步,kd1-wt和kd1-l17r-kt也没有kd1-y11t/l17r-kt或抑肽酶有效。重要的是,在较高浓度下(≥7.5μm),kd1-y11t/l17r-kt类似于抑肽酶恢复了ma和g以及抑制了纤维蛋白溶解。
158、在teg实验中,通过kd1-wt、kd1-l17r-kt、kd1-y11t/l17r-kt和抑肽酶对最大幅度(ma)、剪切弹性模量强度(g)和ly60的浓度依赖性增强进行多重比较分析如图17-19所示。在1μm抑制剂浓度下,ma在对照和除kd1-wt之外的每种kunitz抑制剂之间没有显著差异(图17a)。在2或3μm抑制剂浓度下,与kd1-wt、kd1-l17r-kt和kd1-y11t/l17r-kt相比,抑肽酶对ma的增强具有统计学显著性(p<0.05)(图17b-c)。高于3μm,kd1-l17r-kt、kd1-y11t/l17r-kt和抑肽酶的ma增强在统计学上没有差异;然而,对于kd1-wt,它显著较低(图17d-e)。对于eaca,进行学生t检验以比较对照和测试的每个eaca浓度之间的ma(图17f)。在500μm、1000μm或3000μmeaca浓度下,与对照相比,ma增强具有统计学显著性。
159、值得注意的是,高达3μm,与基于kd1的抑制剂相比,抑肽酶显著增强了g(图18a-c)。令人惊讶的是,在5和7.5μm抑制剂浓度下,与其他抑制剂相比,kd1-y11t/l17r-kt对g的增强显著更高(图18d-e)。这种观察到的kd1-y11t/l17r-kt相对于抑肽酶的凝块剪切强度g的改善可能是由于抑肽酶抑制fxia和激肽释放酶相对于kd1-y11t/l17r-kt基本上没有抑制作用。此外,图19显示了所选浓度下每种抑制剂的ly60的多重比较分析。在1或5μm时,与每种kd1抑制剂相比,抑肽酶在预防纤维蛋白溶解方面明显更好,而kd1-wt不如在所有测试浓度下的每种抑制剂。在2或3μm时,kd1-y11t/l17r-kt和抑肽酶优于kd1-l17r-kt,并且在7.5μm浓度下没有观察到任何抑制剂的ly60。总体而言,看起来在teg实验中,抑肽酶和kd1-y11t/l17r-kt在抑制纤维蛋白溶解方面优于其他抑制剂。
160、3.7.细胞毒性研究
161、在此,我们想要了解与抑肽酶和目前使用的抗纤维蛋白溶解剂eaca和txa相比,kd1-y11t/l17r-kt的潜在毒性。患者通常在接受大手术时经由静脉内注射或在外伤情况下经由外用使用抗纤维蛋白溶解剂进行治疗。因此,我们测试了最有可能暴露于治疗剂量的kd1-y11t/l17r-kt的细胞内皮细胞和皮肤成纤维细胞的细胞毒性。txa在人、大鼠和狗体内的血浆半衰期约为120分钟(48)。两种kd1变体同源物(抑肽酶和艾卡拉肽)中的每个在人中的半衰期也约为120分钟(49,50),而抑肽酶在小鼠、大鼠或狗中的半衰期约为70分钟(51)。每种kd1变体的半衰期是未知的,但可能短,并计划确定该半衰期。由于每种抗纤维蛋白溶解剂在体内的半衰期都短,输注通常在整个外科手术过程中持续进行。因此,治疗持续时间设定为24小时,所选剂量范围包括相当于每种测试药剂临床剂量的约3x。
162、在0.1μm-30μm的整个剂量范围内,与用磷酸盐缓冲液对照处理的细胞相比,用kd1-y11t/l17r-kt或抑肽酶处理huvec24小时的的刃天青测定没有导致细胞存活力的任何显著变化(图11a,vadivel et a1.,j clin med.2020 nov 17;9(11):3684.doi:10.3390/jcm9113684)。用eaca(剂量范围1-60mm)和txa(剂量范围0.2-30mm)处理后获得了相同的结果。细胞存活力在原代人皮肤成纤维细胞中同样没有变化(vadivel et al.,j clinmed.2020 nov 17;9(11):3684.doi:10.3390/jcm9113684中的图11b),表明所测试的抗纤维蛋白溶解剂均未在24小时治疗期间引起可测量的细胞毒性。
163、存活力是细胞毒性的终点。因此,我们检查了由半胱天冬酶激活引起的细胞凋亡的诱导。在用抗纤维蛋白溶解剂处理后的huvec细胞中进行半胱天冬酶3/7测定(vadivelet al.,j clin med.2020 nov 17;9(11):3684.doi:10.3390/jcm9113684中的图11c)。当细胞用两个较高浓度(10mm和30mm)的txa处理时,半胱天冬酶3/7活性显著增加,而在暴露于eaca(20mm和60mm)后,半胱天冬酶3/7活性增加程度较小。与txa和eaca相比,kd1-y11t/l17r-kt和抑肽酶在所有浓度下均未诱导高于基线的半胱天冬酶活性。将紫杉醇作为阳性对照包括在内。在所有剂量中,没有一种抗纤维蛋白溶解剂使原代成纤维细胞中的半胱天冬酶活性高于基线。
164、为了使用不同的测定确认上述结果,我们在huvec细胞和原代成纤维细胞中进行了celltox绿色细胞毒性测定。celltox绿色染料结合dna,只有在膜完整性受损时才会产生荧光染色。在内皮细胞中(vadivel et al.,j clin med.2020nov 17;9(11):3684.doi:10.3390/jcm9113684中的图11d)或在成纤维细胞中(vadivel et al.,j clin med.2020nov 17;9(11):3684.doi:10.3390/jcm9113684中的图11e)用最高剂量(30μm)的kd1-y11t/l17r-kt处理后24小时,没有检测到荧光细胞百分比的显著增加。当用其他测试的抗纤维蛋白溶解剂处理细胞(huvec或成纤维细胞)时,细胞毒性也没有显著增加超过基线。为简洁起见,未显示抑肽酶、txa和eaca的数据。
165、总之,用0.1-30μm kd1-y11t/l17r-kt或抑肽酶对huvec细胞和原代人成纤维细胞进行24小时处理,不会降低存活力、诱导细胞凋亡或显示任何细胞毒性迹象。然而,根据半胱天冬酶3/7活性的增加推断,较高浓度的txa和eaca在huvec细胞中诱导细胞凋亡(细胞死亡)。
166、4.讨论
167、早些时候,基于结构信息和s2′-亚位点特异性,我们设计了来自tfpi-2kd1的73个残基kunitz结构域纤溶酶抑制剂(32)。kd1-wt以相当的亲和力抑制纤溶酶以及pklk、fxia和fviia/tf,而kd1-l17r仅抑制纤溶酶。残基17(bpti编号)从leu到arg的变化使kd1-l17r对纤溶酶具有特异性,并显著降低了pklk和fxia抑制。与目前的60个残基kd1-l17r-kt相比,除了核心kunitz结构域之外,先前表达的kd1-l17r在n端有13个额外的残基(9个来自tfpi-2序列,4个来自iia切割位点)以及在c端有4个残基(vpkv)。尽管这些额外的残基不会干扰kd1-l17r的功能,但它们是柔性的,并且可以从kd1-wt的晶体结构中推断出可能是无序的(36)。因此,表达了新的60个残基kd1-l17r-kt突变体,并表征了其抑制概况。由于60个残基kd1-l17r-kt的活性位点抑制概况与先前表达的73个残基kd1-l17r没有任何变化,因此预计kd1-l17r-kt在减少失血方面非常有效,其在测试的两种小鼠出血模型(肝裂伤和尾部截肢)中可与抑肽酶相当(32,41,52)。
168、73个残基的kd1-l17r在c端有iekvpkv,而缬氨酸可以通过与iia的延长孵育去除(41)。去除c端的val残基会产生c端赖氨酸,这通过抑制纤溶酶活性位点和纤溶酶原激活使kd1-l17r成为纤维蛋白溶解的双重反应性抑制剂(41)。此外,与iia的延长孵育导致具有不同n端残基的异质kd1-l17r群(41)。纤溶酶和kd1-l17r的模型复合物的结构分析表明,将残基tyr11变为thr将有利于纤溶酶抑制。kd1-y11t/l17r-kt中的苏氨酸与纤溶酶的残基q192形成额外的氢键(图9a)。有趣的是,73残基kd1-l17r在c端片段(iekvpkv)含有两个赖氨酸残基,它们中的任何一个都可以作为c端残基。进一步,具有c端iek序列的60个残基kd1-l17r-kt的建模显示它将增强与纤溶酶原和tpa的kringle结构域的相互作用(图9a和b)。不受特定理论或作用机制的束缚,认为与vpk序列相比,iek序列具有两个额外的相互作用,这些相互作用由kunitz结构域的arg57和glu59分别与纤溶酶kringle残基glu151和arg153引起(图9b)。预计tpa的kringle结构域也会发生类似的相互作用。
169、新的大肠杆菌表达的有c端iek序列的kd1-l17r-kt和kd1-y11t/l17r-kt二者均含有his6标签和肠激酶切割序列;然而,肠激酶无法去除这些额外的残基。与73个残基kd1-l17r构建体类似,额外残基的存在不影响kd1-l17r-kt和kd1-y11t/l17r-kt突变体的抑制特性。因此,在巴斯德毕赤酵母中表达60个残基的kd1-y11t/l17r-kt。正如预测的那样,kd1-y11t/l17r-kt与kd1-l17r-kt相比,以增加的亲和力抑制纤溶酶(0.59nm对0.9nm)。进一步,与有c端vpk的kd1-l17r-kcooh(250nm至300nm)(41)相比,有iek c端的60个残基kd1-y11t/l17r-kt以增加的亲和力(35nm至50nm)结合tpa和纤溶酶的kringle结构域(图13)。纤溶酶活性位点抑制的适度增加和对纤溶酶原和tpa的kringle结构域的亲和力的显著提高反映在kd1-y1 1t/l17r-kt在血浆凝块溶解测定中对纤维蛋白溶解的强烈抑制(图14b)和在teg实验中恢复ma、g和ly60(图16b-f)中。
170、巴斯德毕赤酵母中产生的kd1双突变体(kd1-y11t/l17r-kt)是紧凑、均质的,且有效的人源特异性纤溶酶抑制剂。在纤溶酶抑制测定、血浆凝块溶解测定和teg实验中,kd1-y11t/l17r-kt的特性与抑肽酶相当。此外,kd1-y11t/l17r-kt不抑制pklk、fxia和fviia/stf。进一步,kd1-y11t/l17r-kt在原代内皮细胞或皮肤成纤维细胞中不诱导任何可测量的细胞毒性。然而,txa和eaca在较高浓度下引起这些细胞的凋亡,这可以在这些抗纤维蛋白溶解剂的肾清除过程中实现。这些结果与kd1-l17r-kcooh(c端vpk)单突变体一致,kd1-l17r-kcooh不会诱导肾毒性或癫痫发作或任何可检测的在小鼠肾脏中组织病理学变化(32)。在抑肽酶的情况下,其酸性性质和pklk抑制会导致肾活性改变,从而导致肾脏损伤(32,53)。当前的抗纤维蛋白溶解剂eaca和txa通过抑制甘氨酸受体引起癫痫发作(54)。由于赖氨酸类似物不如抑肽酶有效,因此较高剂量的eaca和txa会增加肾衰竭的风险,因为这些试剂在肾小球滤过清除过程中会达到非常高的浓度(55,56)。本公开中发现的kd1y11t/l17r-kt数据令人鼓舞;然而,在考虑用于临床试验之前,需要在合适的动物出血模型中对其进行评估。
171、
172、
173、nhb,正常人血;ma,最大幅度(最大凝块强度);g,剪切强度;ly30,凝块形成后30分钟观察到的溶解百分比;ly60,凝块形成后60分钟观察到的溶解百分比。提供平均值±sd。
174、实施例1参考文献
175、1.kashuk,j.l.;moore,e.e.;sawyer,m.;wohlauer,m.;pezold,m.;barnett,c.;biffl,w.l.;burlew,c.c.;johnson,j.l.;sauaia,a.primary fibrinolysis is integralin the pathogenesis of the acute coagulopathy of trauma.ann.surg.2010,252,434-442.
176、2.raza,i.;davenport,r.;rourke,c.;platton,s.;manson,j.;spoors,c.;khan,s.;de’ath,h.d.;allard,s.;hart,d.p.;pasi,k.j.;hunt,b.j.;stanworth,s.;maccallum,p.k.;brohi,k.the incidence and magnitude of fibrinolytic activationintrauma patients.j.thromb.haemost.2013,11,307-314.
177、3.cardenas,j.c.;wade,c.e.;holcomb,j.b.mechanisms of trauma induced coagulopathy.curr.opin.hematol.2014,21,404-409.
178、4.evans,j.a.;van wessem,k.j.;mcdougall,d.;lee,k.a.;lyons,t.;balogh,z.j.epidemiology of traumatic deaths:comprehensive population-basedassessment.world j.surg.2010,34,158-163.
179、5.oyeniyi,b.t.;fox,e.e.;scerbo,m.;tomasek,j.s.;wade,c.e.;holcomb,j.b.trends in 1029 trauma deaths at a level 1 trauma center:impact of ableeding control bundle of care.injury 2017,48,5-12.
180、6.ker,k.;roberts,i.;shakur,h.;coats,t.j.antifibrinolytic drugs foracute traumatic injury.cochrane database syst.rev.2015,cd004896.
181、7.koster,a.;faraoni,d.;levy,j.h.antifibrinolytic therapy for cardiacsurgery:an update.anesthesiology 2015,123,214-221.
182、8.royston,d.;van haaften,n.;de vooght,p.aprotinin;friend or foe?areview of recent medical literature.eur.j.anaesthesiol.2007,24,6-14.
183、9.mangano,d.t.;tudor,i.c.;dietzel,c.;multicenter study ofperioperative ischemia research group;ischemia research and educationfoundation.the risk associated with aprotinin in cardiacsurgery.n.engl.j.med.2006,354,353-365.
184、10.immer,f.f.;jent,p.;englberger,l.;stalder,m.;gygax,e.;carrel,t.p.;tevaearai,h.t.aprotinin in cardiac surgery:a differem point of view.heartsurg.forum.2008,11,e9-12.
185、11.beierlein,w.;scheule,a.m.;dietrich,w.;ziemer,g.forty years ofclinical aprotinin use:a review of 124 hypersensitivityreactions.ann.thorac.surg.2005,79,741-748.
186、12.fergusson,d.a.;hebert,p.c.;mazer,c.d.;fremes,s.;macadams,c.;murkin,j.m.;teoh,k.;duke,p.c.;arellano,r.;blajchman,m.a.;bussieres,j.s.;cote,d.;karski,j.;martineau,r.;robblee,j.a.;rodger,m.;wells,g.;clinch,j.;pretorius,r.;bart investigators.a comparison ofaprotinin and lysine analoguesin high-risk cardiac surgery.n.engl.j.med.2008,358,2319-2331.
187、13.markus,g.;depasquale,j.l.;wissler,f.c.quantitative determinationof the binding of epsilon-aminocaproic acid to nativeplasminogen.j.biol.chem.1978,253,727-732.
188、14.mathews,i.i.;vanderhoff-hanaver,p.;castellino,f.j.;tulinsky,a.crystal structures of the recombinant kringle 1 domain of human plasminogenin complexes with the ligands epsilon-aminocaproic acid and trans-4-(aminomethyl)cyclohexane-1-carboxvlic acid.biochemistry 1996,35,2567-2576.15.tempe,d.k.;hasija,s.are tranexamic acid and ε-aminocaproic acidadequate substitutes for aprotinin?.ann.card.anaesth.2012,15,4-5.
189、16.martin,k.;gertler,r.;liermanm,h.;mayr,n.p.;macguill,m.;schreiber,c.;vogt,m.;tassani,p.;wiesner,g.switchfrom aprotinin to ε-aminocaproic acid:impact on blood loss,transfusion,and clinical outcome in neonates undergoingcardiac surgery.br.j.anaest.2011,107,934-939.
190、17.martin,k.;knorr,j.;breuer,t.;gertler,r.;macguill,m.;lange,r.;tassani,p.;wiesner,g.seizures after open heart surgery:comparison of epsilon-aminocaproic acid and tranexamic acid.j.cardiothorac.vasc.anesth.2011,25,20-25.
191、18.markland,w.;ley,a.c.;lee,s.w.;ladner,r.c.iterative optimization ofhigh-affinity proteases inhibitors using phage display.1.plasmin.biochemistry1996,35,8045-8057.
192、19.flight,s.m.;johnson,l.a.;du,q.s.;warner,r.l.;trabi,m.;gaffney,p.j.;lavin,m.f.;de jersey,j.;masci,p.p.textilinin-1,analternative anti-bleeding agent to aprotinin:importance of plasmin inhibition in controllingblood loss.br.j.haematol.2009,145,207-211.
193、20.dietrich,w.;nicklisch,s.;koster,a.;spannagl,m.;giersiefen,h.;vande locht,a.cu-2010-a hovel small molecule protease inhibitor withantifibrinolytic and anticoagulant properties.anesthesiology 2009,110,123-130.
194、21.swedberg,j.e.;harris,j.m.plasmin substrate binding sitecooperativity guidesthe design of potent peptide aldehydeinhibitors.biochemistry 2011,50,8454-8462.
195、22.saupe,s.m.;leubner,s.;betz,m.;klebe,g.;steinmetzer, t.developmentof new cyclic plasmin inhibitors with excellent potency andselectivity.j.med.chem.2013,56,820-831.
196、23.al-horani,r.a.;desai,u.r.recent advances on plasmin inhibitors forthe treatment ofibrinolysis-related disorders.med.res.rey.2014;34:1168-216.
197、24.de veer,s.j.;wang,c.k.;harris,j.m.;craik,d.j.;swedberg,j.e.improving the selectivity of engineered protease inhibitors:optimizingthe p2 prime residue using a versatile cyclic peptidelibrary.j.med.chem.2015,58,8257-8268.
198、25.swedberg,j.e.;wu,g.;mahatmanto,t.;durek,t.;caradoc-davies,t.t.;whisstock,j.c.;law,r.;craik,d.j.highly potent and selective plasmininhibitors based on the sunflower trypsin inhibitor-1scaffold attenuatefibrinolysis in plasma.j.med.chem.2019,62,552-560.
199、26.earl,s.t.;masci,p.p.;de jersey,j.;lavin,m.f.;dixon,j.drugdevelopment from australian elapid snake venoms and the venomics pipeline ofcandidates for haemostasis:textilinin-1(q8008),haempatchtm(q8009)and covasetm(v0801).toxicon.2012,59,456-463.
200、27.cheng,l.;pettersen,d.;ohlsson,b.;schell,p.;karle,m.;evertsson,e.;pahlén,s.;jonforsen,m.;plowright,a.t.;j.;fex,t.;thelin,a.;hilgendorf,c.;xue,y.;wahlund,g.;lindberg,w.;larsson,l.o.;gustafsson,d.discovery of the fibrinolysis inhibitor azd6564,acting via interference ofa protein-protein interaction.acsmed.chem.lett.2014,5,538-543.
201、28.jankun,j.;keck,r.;selman,s.h;skrzypczak-jankun,e.systemic ortopical application of plasminogen activator inhibitor with extended half-life(vlhl pai-1)reduces bleeding time and total bloodloss.int.j.mol.med.2010,26,501-504.
202、29.devy,l.;rabbani,s.a.;stochl,m.;ruskowski,m.;mackie,i.;naa,l.;toews,m.;van gool,r.;chen,j.;ley,a.;ladner,r.c.;drahsfield,d.t.;henderikx,p.pegylated dx-1000:pharmacokinetics and antineoplastic activity of aspecific plasmin inhibitor.neoplasia2007,9,927-937.
203、30.bokesch,p.m.;szabo,g.;wojdyga,r.;grocott,h.p.;smith,p.k.;mazer,c.d.;vetticaden,s.;wheeler’a.;levy,j.h.a phase 2prospective,randomized,double-blind trial comparing the efffects oftranexamic acid with ecallantideon blood loss from high-risk cardiac surgery with cardiopulmonary bypass(conserv-2trial).j. thorac.cardiovasc.surg. 2012,143,1022-1029.
204、31.englberger,l.;dietrich,w.;eberle,b.;erdoes,g.;keller,d.;carrel,t.anovel blood-sparing agent in cardiac surgery?first inpatient experiennce withthe synthetic serine protease inhibitor mdco-2010:a phase ii,randomized,double-blind,placebo-controlled study in patients undergoing coronary arterybypass grafting with cardiopulmonary bypass.anesth.analg.2014,119,16-25.
205、32.bajaj,m.s.;ogueli,g.i.;kumar,y.;vadivel,k.;lawson,g.;shanker,s.;schmidt,a.e.;bajaj,s.p.engineering kunitz domain 1(kd1)of human tissue factorpathway inhibitor-2to selectively inhibit fibrinolysis:properties of kd1-l17rvariant.j.biol.chem.2011,286,4329-4340.
206、33.royston,d.;bidstrup,b.d.;taylor,k.m.;sapsford,r.n.aprotinindecreases the need for post-operative blood transfusions in patients havingopen heart surgery.bibl.cardiol.1988,43,73-82.
207、34.bajaj,s.p.;schmidt,a.e.;agah,s.;bajaj,m.s.;padmanabhan,k.highresolution structures of p-aminobenzamidine-and bennzamidine-viia/solubletissue factor:unpredicted conformation of the 192-193peptide bond and mappingof ca2+,mg2+,na+,and zn2+sites infactor viia.j.biol.chem.2006,281,24873-24888.
208、35.sambrook,j.;russel,d.w.molecular cloning:a laboratory mannal, 3rded.,cold spring harbor laboratory press,cold spring harbor,ny,usa,2001.
209、36.schmidt,a.e.;chand,h.s.;cascio,d.;kisiel, w.;bajaj,s.p.crystalstructure of kunitz domain 1(kd1)of tissue factor pathway inhibitor-2 incomplex with trypsin.implications for kd1 specificity ofinhibition.j.biol.chem.2005,280,27832-27838.
210、37.laemmli,u.k.cleavage of structural proteins during assembly ofhead of bacteriophage-t4.nature 1970,227,680-685.
211、38.beith,j.g.in vivo significance ofkinetic constants ofproteinproteinase inhibitors.biochem.med.1984,32,387-397.
212、39.morrison,j.f.;walsh,c.t.the behavior and significance ofslowbinding enzyme inhibitors.adv.enzymol.relat.areas mol.biol.1988,61,201-301.
213、40.sperzel, m.;huetter,j.evaluation of aprotinin andtranexamic acidin different in vitro and in vivo models of fibrinolysis,coagulation andthrombus formation.j.thromb.haemost.2007,5,2113-2118.
214、41.kumar,y.;vadivel,k.;schmidt,a.e.;ogueli,g.i.;ponnuraj,s.m.;rannulu,n.;loo,j.a.;bajaj,m.s.;bajaj,s.p.decoy plasminogen receptorcontaining a selective kunitz-inhibitory domain.biochemistry 2014,53,505-517.
215、42.chandler,w.l.the thromboelastography and the thromboelastograph techniqne.semin.thromb.hemost.1995,21,(suppl 4):1.
216、43.parry,m.a.a.;fernandez-catalan,c.;bergner,a.;huber,r.;hopfner,k.p.;schlott,b.;guhrs,k.h.;bode,w.(1998)the ternary microplasmin-staphylokinase-microplasmin complex is a proteinase-cofactor-substratecomplex in action,nat.struct.biol.1998,5,917-923.
217、44.bajaj,m.s.;birktoft,j.j.;steer,s.a.;bajaj,s.p.structure andbiology of tissue factor pathway inhibitor,thromb.haemostasis 2001,86,959-972.
218、45.eswar,n.;marti-renom,m.a.;webb,b.;madhusudhan,m.s.;eramian,d.;shen,m.;pieper,u.;sali,a.(2006)comparative protein structure modeling withmodeller.current protocols in bioinformatics,john wiley&sohs,inc.,supplement15,5.6.1-5.6.30.
219、46.case,d.a.;betz,r.m.;botello-smith,w.;cerutti,d.s.;cheatham iii,t.e.;darden,t.a.;duke,r.e.;giese,t.j.;gohlke,h.;goetz,a.w.;homeyer,n.;izadi,s.;janowski,p.;kaus,j.;kovalenko,a.;lee,t.s.;legrand,s.;li,p.;lin,c.;luchko,t.;luo,r.;madej,b.;mermelstein,d.;merz,k.m.;monard,g.;nguyen,h.;nguyen,h.t.;omelyan,i.;onufriev,a.;roe,d.r.;roitberg,a.;sagui,c.;simmerling,c.l.;swails,j.;walker,r.c.;wang,j.;wolf,r.m.;wu,x.;xiao,l.;york,d.m.;kollman,p.a.amber,university of california,san francisco,usa,2018.
220、47.light,a.;fonseca,p.the preparation and properties of the catalyticsubunit of bovine enterokinase.j.biol.chem.1984,259,13195-13198.
221、48.australian public assessment report for tranexamicacid.cyklokapran.https://www.tga.gov.au/sites/default/files/auspar-cyklokapron.pdf accessed 4 october 2020.
222、49.trasylol.
223、http://www.accessdata.fda.gov/drugsatfda_docs/label/2006/020304s022lbl.pdf accessed 4 october 2020.
224、50.farkas,h.;varga,l.(2011)ecallantide is a novel treatment forattacks of hereditary angioedema due to c1 inhibitor deficiency.clin.cosmet.investig.dermatol.2011, 4,61-68.
225、51.markwardt,f.naturally occurring inhibitors offibrinolysis.fibrinolytics and anti-fibrinolytics,ed.f.markwardt,springer-verlag,1978,pages 487-509.
226、52.vadivel,k.;kumar,y.;ogueli,g.i.;ponnuraj,s.m.;wongkongkathep,p.;loo,j.a.;bajaj,m.s.;bajajsp.s2′-subsite variations between human and mouseenzymes(plasmin,factor xia,kallikrein)elucidate inhibition differences bytissue factor pathway inhibitor-2domain1-wild-type,leu17arg-mutant andaprotinin.j.thromb.haemost.2016,14,2509-2523.
227、53.seto,s.;kher,v.;scicli,a.g.;beierwaltes,w.h.;carretero,o.a.(1983).the effect of aprotinin(a serine protease inhibitor)on renal function andrenin release.hypertension,1983,5,893-899.
228、54.lecker,i.;wang,d.s.;romaschin,a.d.;peterson,m.;mazer,c.d.;orser,b.a.tranexamic acid concentrations associated withhuman seizures inhibitglycine receptors.j.clin.invest.2012,122,4654-4666.
229、55.pilbrant,a.;schannong,m.;vessman,j.(1981).pharmacokinetics andbioavailability of tranexamic acid.eur.j. clin.pharma.1981,20;65-72.
230、56.frederiksen,m.c.;bowsher,d.j.;ruo,t.i.;henthom,t.k.;ts′ao,c.h.;green,d.;atkinson,a.j.jr.kinetics of epsilon-aminocaproic acid distribution,elimination,and antifibrinolytic effects in normal subjects.clin.pharmacol.ther.1984,35,387-393.
231、实施例2:包含60个残基的kd1-y11t/r15k/l17r三重突变体(rhukd1-tm)的新的纤溶酶特异性kunitz抑制剂多肽,其具有意想不到的功能特征
232、先前研究的抗纤维蛋白溶解多肽变体包括2型人组织因子途径抑制剂的kunitz结构域1(kd1)的单突变体和双突变体(kd1l17r、kd1y11t/l17r)(参见例如美国专利号7,585,842和美国专利公开号20080026998和20140288),其成功地防止了两种小鼠损伤模型(肝脏撕裂laceration模型和尾部切断模型)的失血。然而,这些多肽变体的效力具有局限性,例如,与使用抑肽酶的类似研究相比,它们在与组织纤溶酶原激活剂长时间孵育后在血浆凝块溶解测定中表现出稍微降低的活性。
233、为了在血浆凝块溶解测定中增加此类变体的效力,进一步制备和研究了2型人组织因子途径抑制剂变体的kunitz结构域1(kd1)。作为这些研究的一部分,开发了具有独特氨基酸突变群的截短三重突变体(kd1y11t/r15k/l17r)。
234、该变体包含以下序列:
235、naeicllpldtgpckarllryyydrytqscrqflyggcegnannfytweacddacwriek(seq idno:1)。
236、以下提供了单、双和三重kd1突变体的序列比较及它们的特性。
237、1)人tfpi-2的kunitz结构域1(kd1)的编号系统:
238、
239、本文公开的kd1三重突变体(60个残基,具有序列nh2-1naei…iek60-cooh,bpti编号)在毕赤酵母中表达并纯化,显示出7.1kda的分子量(图1)。如上所述,该变体被发现是强大的纤溶酶抑制剂(图2),类似于抑肽酶,而没有抑制其他凝血丝氨酸蛋白酶(包括激肽释放酶)的副作用(图2)。因此,与作为广泛特异性蛋白酶抑制剂的抑肽酶相比,本文公开的kd1三重突变体是人源的并且对抑制纤溶酶非常特异。
240、表4-6中呈现的数据说明了本文公开的2型人组织因子途径抑制剂的kunitz结构域1(kd1)的60个残基三重突变体多肽变体的某些意想不到的药代动力学特性和意想不到特性的组合。
241、表4:kd1y11t/r15k/l17r-kt对纤溶酶和各种凝血因子的抑制作用的ki值
242、
243、*kd1tm,kd1y11t/r15k/l17r-kt;kd1dm,kd1-y11t/l17r-kt;kd1sm,kd1-l17r。
244、使用紧密结合等式计算每个抑制剂的ki值。与ki为18nm的抑肽酶相比,kd1tm对激肽释放酶的抑制作用直到25μm才观察到。抑肽酶对激肽释放酶的抑制与肾脏损伤相关。赖氨酸类似物氨甲环酸(txa)和ε-氨基己酸(eaca)通过不同的机制发挥作用,不抑制纤溶酶活性位点或其他蛋白酶。
245、表5和6:人tfpi-2kd1突变体与tpa和纤溶酶原的kringle结构域结合
246、从表面等离子体共振实验获得的结合常数
247、表5:有c端iekvpk序列的rhukd1突变体
248、 <![cdata[k<sub>on</sub>(m<sup>-1</sup>s<sup>-1</sup>)]]> <![cdata[k<sub>off</sub>(s<sup>-1</sup>)]]> <![cdata[k<sub>d</sub>(nm)]]> tpa <![cdata[0.91±0.2x10<sup>3</sup>]]> <![cdata[1.9±0.3x10<sup>-4</sup>]]> 210±20 glu-纤溶酶原 <![cdata[1.1±0.4x10<sup>3</sup>]]> <![cdata[3.1±0.8x10-<sup>4</sup>]]> 280±30
249、表6:有c端iek序列的rhukd1突变体
250、 <![cdata[k<sub>0n</sub>(m<sup>-1</sup>s<sup>-1</sup>)]]> <![cdata[k<sub>off</sub>(s<sup>-1</sup>)]]> <![cdata[k<sub>d</sub>(nm)]]> tpa <![cdata[2.91±0.4x10<sup>3</sup>]]> <![cdata[1.05±0.7x10<sup>-4</sup>]]> 35.4±5 dip-δ纤溶酶 <![cdata[1.49±0.3x10<sup>3</sup>]]> <![cdata[7.13±0.9x10<sup>-5</sup>]]> 47.6±7
251、dip-δ纤溶酶:活性位点抑制的纤溶酶仅含有纤溶酶蛋白酶结构城和kringle1结构域
252、如表4-6中提供的数据和图9中的示意图所示,已发现2型人组织因子途径抑制剂的kunitz结构域1(kd1)的这种60个残基三重突变体多肽变体具有非常理想的药代动力学概况。与常规使用但有问题的抗纤维蛋白溶解剂抑肽酶相比,该概况包括更强的抑制纤溶酶活性的能力。此外,虽然这种新的多肽变体的纤溶酶抑制活性比抑肽酶的纤溶酶抑制活性更有利,但本文公开的多肽变体进一步避免了抑肽酶和相关分子观察到的某些不良副作用。在其一个例子中,观察到本文公开的多肽变体对其他凝血丝氨酸蛋白酶,如激肽释放酶、因子xia和因子viia/组织因子,表现出最小的抑制活性。
253、如上所述,本文公开的60个残基多肽变体包括独特的氨基酸残基群,其包含c-端赖氨酸结构/部分。不受特定理论或作用机制的束缚,该c-端结构看起来通过促进60个残基多肽变体以抑制纤溶酶原与纤维蛋白凝块结合的方式与纤溶酶或纤溶酶原经由其kringle结构域结合而起作用。本文公开的多肽变体进一步包括一组三个氨基酸突变(“kd1y11t/r15k/l17r”),包括15位的赖氨酸氨基酸取代。令人惊讶的是,这种包含c端赖氨酸的y11t/r15k/l17r三重突变体与仅具有双重突变y11t/l17r的对照60个残基多肽变体相比,观察到其抑制纤溶酶的效力高4至5倍(参见例如图2和表4-6中显示的数据)。不受特定理论或作用机制的束缚,这种在15位具有赖氨酸氨基酸取代的三重突变体看起来通过促进这种变体多肽与纤溶酶中残基asp189和ser190的相互作用来发挥作用。
254、与仅具有双重突变y11t/l17r的对照60个残基多肽变体相比,该60个残基y11t/r15k/l17r三重突变体多肽进一步表现出至少弱10倍的激肽释放酶、因子xia和因子viia/组织因子抑制,这是一种会限制不需要的副作用如在抑肽酶观察到的副作用的功能概况。野生型2型人组织因子途径抑制剂分子中第15位(bpti编号)的arg残基对于抑制因子xia和激肽释放酶很重要。然而,由于因子xia和激肽释放酶在190位具有丙氨酸残基,本文公开的kunitz结构域1多肽抑制剂中第15位的赖氨酸显然不能与因子xia和激肽释放酶中的ala190相互作用。因此,这些在该位置具有赖氨酸的60个残基的多肽是因子xia和激肽释放酶活性的极弱抑制剂。
255、由于上述原因,本文公开的60个残基变体多肽表现出令人惊讶和非常理想的药代动力学/材料概况,包括例如强烈结合纤溶酶的能力,同时避免与该技术中的类似抑制分子相关的某些副作用。此类功能特性使这些多肽最适合用作体内治疗剂。本文所公开的多肽进一步具有许多其他需要的特性。例如,60个残基变体多肽显示与组织纤溶酶原激活剂(tpa)结合并抑制其与纤维蛋白凝块的结合,从而减弱凝血位点的纤溶酶原激活。
256、以下部分讨论了60个残基变体多肽的其它方面。
257、与kd1y11t/r15k/l17r-kt的相互作用建模
258、在图9a中,显示了kd1y11t/r15k/l17r-kt与纤溶酶相互作用的模拟复合物。子部分(a)显示了kd1y11t/r15k/l17r-kt与纤溶酶蛋白酶结构域的模拟相互作用。描绘了纤溶酶蛋白酶结构域的静电表面和kdly11t/r15k/l17r-kt(浅绿色)的卡通表示。kd1y11t/r15k/l17r-kt的p1(lys15)、p5(thr11)和p2′(arg17)残基与纤溶酶相互作用以棒状表示形式显示。在静电表面,蓝色代表正电荷,红色代表负电荷,白色代表中性电荷。子部分(b)显示了kd1y11t/r15k/l17r-kt与纤溶酶kringle结构域的模拟相互作用。描述了纤溶酶原kringle结构域1的静电表面和kd1y11t/r15k/l17r-kt(浅绿色)的卡通表示。在kringle结构域和kd1y11t/r15k/l17r-kt之间形成氢键和盐桥(显示为虚线)的残基以棒状表示形式显示。kringle结构域的碳原子显示为绿色,kd1y11t/r15k/l17r-kt的碳原子显示为黄色。与子部分(a)中一样,氧原子以红色显示,氮原子以蓝色显示。kd1y11t/r15k/l17r-kt残基用后缀i标记。在静电表面,蓝色代表正电荷,红色代表负电荷,白色代表中性电荷。
259、应注意的是纤溶酶中残基190的ser与rhukd1-tm(kd1y11t/r15k/l17r-kt)中的赖氨酸15相互作用,这对于激肽释放酶或因子xia中的ala 190是不可能的。因此,纤溶酶活性位点对纤溶酶来说亲和力增加,对激肽释放酶和因子xia来说亲和力降低。此外,不受具体理论或作用机制的束缚,kd1y11t/r15k/l17r-kt多肽变体中的iek c端看起来比具有vpk c端序列的多肽具有更多的与纤溶酶的相互作用,因此对纤溶酶kringle结构域具有更高的亲和力。此外,类似的kd1y11t/r15k/l17r-kt多肽变体与组织纤溶酶原激活剂的kringle结构域发生相互作用。
260、图9a提供了kd1y11t/r15k/l17r-kt多肽变体观察到的纤溶酶抑制水平的信息,包括为什么与具有vpkc端基序的多肽相比,iekc端基序更好地结合纤溶酶kringle结构域。这个发现得到了对73个残基长(加上来自凝血酶溶解的4个残基)kuniz结构域(该结构域是公开于美国专利公开号20080026998中的在第15位发生arg到lys(与在第24位相同)以及vpkv末端序列的现有技术的构建体)的研究的支持。该现有技术构建体在多肽的c末端没有lys氨基酸,并且不与纤溶酶kringle结构域结合。此外,在这个特定的73个残基的现有技术分子中n端的9个残基和c端的4个残基是溶剂暴露的并且非常无序,这可能损害该现有技术突变体结合能力的方面。相比之下,当前的三重突变体构建体为60个残基长((kd1y11t/r15k/l17r-kt),其具有c端iek,并且观察到以非常高的亲和力(~40nm)与kringle结构域结合。此外,如以下部分所讨论的,本文公开的2型人组织因子途径抑制剂的kunitz结构域1(kd1)的多肽变体(seq id no:1)表现出非常理想的稳定性概况。
261、稳定性研究
262、本文公开的2型人组织因子途径抑制剂的kunitz结构域1(kd1)的多肽变体表现出意想不到的和理想的稳定性概况。例如,在本发明的某些实施方案中,本文公开的60个残基的多肽变体配置在组合物中,其中当该多肽组合物在包含0.1mg/ml牛血清白蛋白(bsa)和2mm钙的tris缓冲盐水(tbs)中于37℃孵育至少1周时多肽的纤溶酶抑制常数(ki)变化小于10%(或小于5%)。
263、在稳定性研究中,将kd1y11t/r15k/l17r-kt样品(0.59mg/ml)在4℃、室温和37℃下保存一周,每天研究其纤溶酶抑制作用。rhukd1-tm看起来是稳定的并且其纤溶酶抑制特性没有改变。类似于抑肽酶的rhukd1-tm突变体是缓慢的紧密结合的纤溶酶抑制剂。反应在tbs/bsa和2mm钙中进行。在96孔微量滴定板中,人纤溶酶与各种浓度的rhukd1-tm在室温下孵育1小时。然后将纤溶酶的合成底物添加至1km的最终浓度,并在动态酶标仪(moleculardevices)中测量残留的酰胺分解活性。在下表7中,表观抑制常数ki*是使用非线性回归数据分析程序确定的。使用紧密结合抑制剂的等式分析数据,并通过校正底物浓度的影响获得ki值。
264、表7:三重突变体(kd1y11t/r15k/l17r-kt)稳定性数据
265、
266、从该表中的数据可以清楚看出rhukd1-tm非常稳定。
267、kd1y11t/r15kl17r-kt(rhukd1-tm)和抑肽酶抑制纤溶酶的平衡解离常数(ki)
268、rhukd1-tm和抑肽酶抑制纤溶酶的平衡解离常数(ki)是使用缓慢紧密结合抑制方程等式的(10,11)。rhukd1-tm与纤溶酶结合的ki值在不同实验中范围为50至150皮摩尔,相比抑肽酶的ki值为400至500皮摩尔(图2)。此外,c端60个赖氨酸应允许rhukd1-tm与纤溶酶原/纤溶酶和组织纤溶酶原激活剂(tpa)的kringle结构域结合,并抑制它们与纤维蛋白凝块结合。rhukd1-tm的这一特性进一步抑制纤溶酶引起的纤维蛋白溶解。因此,rhukd1-tm(kd1y11t/r15k/l17r-kt)是非常有效且独特的纤维蛋白溶解抑制剂。
269、在kd1双突变体(y11t/l17r)中将arg15更改为lys有效地与纤溶酶的s1位点asp189相互作用,其中包括纤溶酶的ser190与rhukd1-tm的lys15的相互作用,如胰蛋白酶与抑肽酶的结构中所定义(12)。lys15的此类相互作用不可能与具有ala190(13,14)而不是ser的激肽释放酶或因子xia和因子xa发生。因此,kd1y11t/r15k/l17r-kt对这三种酶的抑制极差(图2),这是在患者中使用的理想结果。虽然因子viia具有ser190,但与单突变体或双突变体类似,三重突变体(图2)对它的抑制很差。
270、rhukd1-tm和抑肽酶对人正常混合血浆中纤维蛋白溶解的影响。
271、进行实验以比较三重突变体kd1y11t/r15k/l17r-kt和抑肽酶抑制tpa诱导的血浆凝块纤维蛋白溶解的有效性。这些数据针对三重突变体显示在图3a中和针对抑肽酶显示在图3b中。将凝血酶(iia)添加到人正常混合血浆(npp)中会导致纤维蛋白形成,这反映在od405的增加上(黑色曲线,图3a和3b)。同时添加tpa导致初始凝块形成,随后由tpa介导的纤溶酶原转化为纤溶酶诱导纤维蛋白溶解(带闭合圆圈的黑色曲线,图3a和3b);在每种情况下,纤维蛋白溶解的中点约为8分钟。两种抗纤维蛋白溶解剂均以剂量依赖性方式抑制纤维蛋白溶解。kd1y11t/r15k/l17r-kt(三重突变体)和抑肽酶在0.5μm时将纤维蛋白溶解中点增加至约15分钟,并且在3μm时,两种抑制剂都完全阻止了tpa诱导的纤维蛋白溶解(图3a和3b中的洋红色曲线)。重要的是,在没有tpa的情况下添加3μm的每种抑制剂对凝块形成或纤维蛋白溶解没有影响(棕色曲线,图3a和3b)。因此,kd1y11t/r15k/l17r-kt和抑肽酶在较长时间内防止tpa诱导的血浆凝块溶解方面基本上是等效的。
272、抗纤维蛋白溶解剂对原代细胞存活力的影响
273、检测了每种抗纤维蛋白溶解剂rhukd16tm、抑肽酶、eaca和txa对人脐静脉内皮细胞(huvec)和皮肤成纤维细胞存活力的影响。huvec的数据显示在图4中,皮肤成纤维细胞的数据显示在图5中。在24小时内,所测试的抗纤维蛋白溶解剂均未对所测试的两种细胞类型的存活力产生可检测的影响。请注意,活细胞在胞质溶质内保持还原环境。活细胞将底物刃天青还原为荧光试卤灵,荧光与细胞数量成比例。无论细胞状态如何,底物刃天青都具有细胞渗透性(进入细胞)。因此,该测定没有检测到任何细胞凋亡或坏死。
274、抗纤维蛋白溶解剂对细胞凋亡的影响
275、细胞凋亡,有时称为“细胞自杀”,是正常的、程序化的细胞自我毁灭过程。在细胞凋亡过程中,细胞收缩并远离其邻居。半胱天冬酶3和7是仅在细胞发生凋亡时才被激活的蛋白酶。在本文使用的测定中,特定底物被半胱天冬酶3和半胱天冬酶7转化为萤光素酶的底物。然后萤光素酶产生发光信号。发光与半胱天冬酶活性成比例。在细胞凋亡过程中,细胞的膜完整性保持完整。在所用的测定中,eaca和txa诱导huvec中的半胱天冬酶活性增加,但不会显著增加成纤维细胞中的半胱天冬酶活性(图6)。因此,eaca和txa可诱导在体内血管内衬的内皮细胞中程序性细胞凋亡。在使用的条件下,rhukd1-tm和抑肽酶(bpti)不会在细胞huvec和皮肤成纤维细胞中诱导细胞凋亡(图6)。
276、抗纤维蛋白溶解剂对细胞毒性的影响
277、这里使用的细胞毒性绿色测定测量由于细胞毒性而发生的膜完整性变化。花青染料不能通过完整的细胞膜进入细胞。如果膜完整性受损,染料会进入细胞并染色dna,从而产生荧光信号。因此,观察到的荧光与细胞毒性成正比。在所用的测定中,txa在huvec和皮肤成纤维细胞中均引起毒性(图7和8),而eaca的显著毒性仅在皮肤成纤维细胞中观察到。在测试条件下,rhukd1-tm和抑肽酶(trasylol,bpti)不会在这些细胞中诱导毒性。
278、血栓弹性成像实验
279、不同浓度的kd1-y11t/r15k/l17r-kcooh(kd1tm)对纤维蛋白溶解的影响使用teg5000血栓弹性成像(haemonetics corp,braintree,ma,usa)通过血栓弹性成像(teg)进行评估。每个凝块形成/溶解测定包含300μl柠檬酸化的全血、凝血酶(0.15μm,最终浓度)、tpa(2nm,最终浓度)、cacl2(10mm,最终浓度)和tbs/bsa(50mmtris-hcl,100mmnacl,含有0.1mg牛血清白蛋白/ml)中不同浓度的kd1tm,使最终体积达到360μl。最后添加凝血酶、tpa和cacl2以同时启动凝血和纤维蛋白溶解。当tpa的浓度导致纤溶酶的产生以在90分钟内几乎完全溶解凝块,则选择该tpa的浓度,允许监测纤溶酶抑制剂的作用。每个实验在达到最大幅度后至少60分钟进行,以确定ly60值。teg分析软件(版本4.2.3;haemoneticscorporation,braintree,ma,usa)用于计算凝块起始时间(r)、最大凝块强度(最大幅度(ma),其与剪切弹性模量强度g直接相关)和ma后60分钟的溶解百分比(ly60)。
280、进行血栓弹性成像实验以评估kd1tm对通过在通过添加凝血酶和cacl2全血中形成的凝块的tpa诱导溶解的影响。这些数据显示在图20中并总结在表8中。图20显示了不同浓度的kd1 tm对使用tpa启动的凝块溶解的teg迹线。在没有tpa的情况下,最大幅度(ma)达到45.5mm,剪切弹性模量强度g为4174dyn/cm2,并且在180分钟时未检测到凝块溶解(ly60<0.1%)(曲线1)。曲线2说明了在没有抑制剂的情况下存在2nm tpa时凝块形成和溶解的teg迹线。曲线3和4说明了在存在2nm tpa的情况下,不同浓度(2μm和4μm)的kd1tm对凝块形成和溶解的teg迹线。数据表明kd1tm以浓度依赖性方式改进了凝块硬度(ma)和剪切强度(g)并抑制了纤维蛋白溶解(表8)。值得注意的是,在4μmkd1tm时,凝块强度ma改进到98%(44.2mm),g改进到96%(4011dyn/cm2)(曲线4)。进一步,与对照相比,没有观察到ly30(ly30,34.6%,曲线2)。类似地,与对照(ly6065%)相比,在2μm和4μm浓度的kd1tm下仅观察到3.7%或1.7%的ly60。重要的是,teg数据表明kd1tm有效地恢复了tpa诱导的纤维蛋白溶解中的ma和g。
281、表8.kd1tm对teg参数的影响。
282、
283、kd1tm:kd1-y11t/r15k/l17r-kcooh
284、本文提供的公开内容得出许多结论,包括以下内容:
285、kd1y11t/r15k/l17r-kt(rhukd1-tm)在抑制纤溶酶的活性位点方面优于kd1-l17r-kcooh(kd1sm)和kd1y11t/l17r-kt(kd1dm),与抑肽酶(trasylol)相当。rhukd1-tm是60个残基的kunitz结构域,以naeic开头,有c端iek(bpti编号)。此外,它不像之前的单突变体(kd1-l17r-kcooh)那样在n端有额外的9个残基。单突变体在c端也有三个额外的残基,以iekvpk结尾。单突变体的缺点是两个疏水残基val和pro降低了它的溶解度。rhukd1-tm是高度可溶的。rhukd1-tm在抑制纤溶酶和血浆凝块溶解测定方面也优于kd1sm和kd1dm,并且在较长的孵育期内与抑肽酶相当。进一步,与单突变体(kd1sm)和双突变体(kd1dm)相比,三重突变体(rhukd1-tm)是激肽释放酶、因子xia、因子xa和因子viia/组织因子的非常弱的抑制剂。
286、大量研究(例如图4-8)表明rhukd1-tm不会对huvec或成纤维细胞以及可能的其他细胞造成损害。通过肾脏清除的eaca和txa可能在肾脏中具有高浓度并导致细胞损伤。因为观察到txa和eaca的副作用,这可能是导致肾功能衰竭的原因。
287、如上所述,已发现rhukd1-tm(seq id no:1)是极好的纤溶酶抑制剂,同时是激肽释放酶和因子xia的极差的抑制剂(并且没有抗凝血活性)。这与源自牛的,由于其对激肽释放酶的抑制而导致肾脏损伤的抑肽酶相反。进一步,两种抑制激肽释放酶的抑制剂(7,8)在iii期心脏搭桥手术试验中非常失败。因此,rhukd1-tm对激肽释放酶和因子xia的极差抑制是所公开的多肽的非常有利的特性。因此,已发现本文公开的60个残基变体多肽具有一系列出乎意料的材料特性,这些特性满足了人们公认的、持续存在的且未被其他人解决的长期需求。
288、本文提及的所有出版物(例如上面按照数字列出的那些,美国专利号8,993,719,美国专利公开号20040126856,20040110688,20080026998,20090018069和20140288000,以及vadivel et a1.,j clin med.2020 nov 17;9(11):3684.doi:10.3390/jcm9113684)通过引用并入本文以公开和描述与引用的出版物相关的方面、方法和/或材料。
- 还没有人留言评论。精彩留言会获得点赞!