吡啶-嘧啶胺-苯并咪唑衍生物及其制备方法和用途
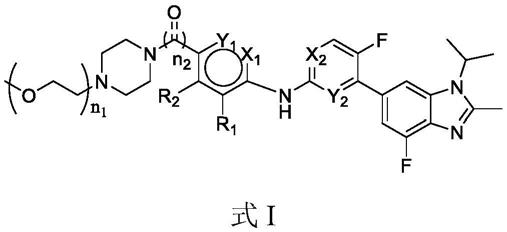
1.本发明属于医药领域,具体涉及吡啶-嘧啶胺-苯并咪唑衍生物及其制备方法和用途。
背景技术:
2.恶性肿瘤具有较高的致死率,并且其发病率逐年增加,已经超过心脑血管类疾病,成为人类健康的第一大杀手。cdk4/6即细胞周期蛋白依赖性激酶4和6,是人体细胞分裂增殖周期的关键条件蛋白,为er、pi3k/mtor等多条促生长信号通路的共同下游靶点。
3.cdk4/6通过cyclin d-cdk4/6-rb通路对细胞周期进行调控。在细胞増殖过程中,cdk4/6与细胞周期素d(cyclin d)形成复合物,能够使视网膜母细胞瘤蛋白(rb)磷酸化。rb一旦发生磷酸化,可使与之紧密结合的转录因子e2f释放。e2f激活进一步推动细胞周期从g1期进入到s期,进入细胞增殖周期。
4.研究发现,几乎所有恶性肿瘤中都有cyclin d-cdk4/6-rb这一通路的失调。表现为cdk4/6过度活跃,这一通路持续激活,最终促使细胞生长失控,促使癌细胞增殖扩散。而抑制cdk4/6,使肿瘤细胞无法形成cyclin d-ck4/6复合物,将细胞周期阻滞于生长期,从而达到抑制肿瘤增殖的目的。因此,以新型小分子抑制剂靶向抑制cdk4/6是一种潜在的治疗方法。
技术实现要素:
5.本发明的目的是提供吡啶-嘧啶胺-苯并咪唑衍生物及其制备方法和用途。
6.本发明提供了式i所示的化合物、或其盐、或其立体异构体、或其溶剂合物、或其水合物、或其前药:
[0007][0008]
其中,
[0009]
n1选自1~3的整数;
[0010]
n2选自0~5的整数;
[0011]
虚线为键或无;
[0012]
x1、y1分别独立选自n或cr3;
[0013]
x2、y2分别独立选自n或cr4;
[0014]
r1、r2分别独立选自氢、c1~c8烷基、c1~c8烷氧基、卤素、羟基、羧基、氨基、硝基;
[0015]
每个r3分别独立选自氢、c1~c8烷基、c1~c8烷氧基、卤素、羟基、羧基、氨基、硝基;
[0016]
每个r4分别独立选自氢、c1~c8烷基、c1~c8烷氧基、卤素、羟基、羧基、氨基、硝基。
[0017]
进一步地,
[0018]
n1选自1~3的整数;
[0019]
n2选自0或1;
[0020]
虚线为键或无;
[0021]
x1、y1分别独立选自n或cr3;
[0022]
x2、y2分别独立选自n或cr4;
[0023]
r1、r2分别独立选自氢、c1~c3烷基、c1~c3烷氧基、卤素;
[0024]
每个r3分别独立选自氢、c1~c3烷基、c1~c3烷氧基、卤素;
[0025]
每个r4分别独立选自氢、c1~c3烷基、c1~c3烷氧基、卤素。
[0026]
进一步地,所述化合物为式ii所示:
[0027][0028]
其中,
[0029]
n1选自1~3的整数;
[0030]
n2选自0~5的整数;
[0031]
x1、y1分别独立选自n或cr3;
[0032]
x2、y2分别独立选自n或cr4;
[0033]
r1、r2分别独立选自氢、c1~c8烷基、c1~c8烷氧基、卤素、羟基、羧基、氨基、硝基;
[0034]
每个r3分别独立选自氢、c1~c8烷基、c1~c8烷氧基、卤素、羟基、羧基、氨基、硝基;
[0035]
每个r4分别独立选自氢、c1~c8烷基、c1~c8烷氧基、卤素、羟基、羧基、氨基、硝基;
[0036]
优选地,
[0037]
n1选自1~3的整数;
[0038]
n2选自0或1;
[0039]
x1、y1分别独立选自n或cr3;
[0040]
x2、y2分别独立选自n或cr4;
[0041]
r1、r2分别独立选自氢、c1~c3烷基、c1~c3烷氧基、卤素;
[0042]
每个r3分别独立选自氢、c1~c3烷基、c1~c3烷氧基、卤素;
[0043]
每个r4分别独立选自氢、c1~c3烷基、c1~c3烷氧基、卤素。
[0044]
进一步地,所述化合物为式iii所示:
[0045][0046]
其中,
[0047]
n1选自1~3的整数;
[0048]
n2选自0~5的整数;
[0049]
x1、y1分别独立选自n或cr3;
[0050]
x2、y2分别独立选自n或cr4;
[0051]
r1、r2分别独立选自氢、c1~c8烷基、c1~c8烷氧基、卤素、羟基、羧基、氨基、硝基;
[0052]
每个r3分别独立选自氢、c1~c8烷基、c1~c8烷氧基、卤素、羟基、羧基、氨基、硝基;
[0053]
每个r4分别独立选自氢、c1~c8烷基、c1~c8烷氧基、卤素、羟基、羧基、氨基、硝基;
[0054]
优选地,
[0055]
n1选自1~3的整数;
[0056]
n2选自0或1;
[0057]
x1、y1分别独立选自n或cr3;
[0058]
x2、y2分别独立选自n或cr4;
[0059]
r1、r2分别独立选自氢、c1~c3烷基、c1~c3烷氧基、卤素;
[0060]
每个r3分别独立选自氢、c1~c3烷基、c1~c3烷氧基、卤素;
[0061]
每个r4分别独立选自氢、c1~c3烷基、c1~c3烷氧基、卤素。
[0062]
进一步地,所述化合物为式iv所示:
[0063][0064]
其中,
[0065]
n1选自1~3的整数;
[0066]
n2选自0~5的整数;
[0067]
虚线为键或无;
[0068]
x1、y1分别独立选自n或cr3;
[0069]
r1、r2分别独立选自氢、c1~c8烷基、c1~c8烷氧基、卤素、羟基、羧基、氨基、硝基;
[0070]
每个r3分别独立选自氢、c1~c8烷基、c1~c8烷氧基、卤素、羟基、羧基、氨基、硝基;
[0071]
优选地,
[0072]
n1选自1~3的整数;
[0073]
n2选自0或1;
[0074]
虚线为键或无;
[0075]
x1、y1分别独立选自n或cr3;
[0076]
r1、r2分别独立选自氢、c1~c3烷基、c1~c3烷氧基、卤素;
[0077]
每个r3分别独立选自氢、c1~c3烷基、c1~c3烷氧基、卤素。
[0078]
进一步地,所述化合物为式v所示:
[0079][0080]
其中,
[0081]
n1选自1~3的整数;
[0082]
n2选自0~5的整数;
[0083]
虚线为键或无;
[0084]
x1、y1分别独立选自n或cr3;
[0085]
每个r3分别独立选自氢、c1~c8烷基、c1~c8烷氧基、卤素、羟基、羧基、氨基、硝基;
[0086]
优选地,
[0087]
n1选自1~3的整数;
[0088]
n2选自0或1;
[0089]
虚线为键或无;
[0090]
x1、y1分别独立选自n或cr3;
[0091]
每个r3分别独立选自氢、c1~c3烷基、c1~c3烷氧基、卤素。
[0092]
进一步地,所述化合物为式va所示:
[0093][0094]
其中,
[0095]
n1选自1~3的整数;
[0096]
n2选自0或1;
[0097]
x1、y1分别独立选自n或cr3;
[0098]
每个r3分别独立选自氢、c1~c3烷基、c1~c3烷氧基、卤素;
[0099]
或者,所述化合物为式vb所示:
[0100][0101]
其中,
[0102]
n1选自1~3的整数;
[0103]
x1、y1分别独立选自n或cr3;
[0104]
每个r3分别独立选自氢、c1~c3烷基、c1~c3烷氧基、卤素。
[0105]
进一步地,所述化合物为如下化合物之一:
[0106]
[0107]
[0108][0109]
本发明还提供了前述的化合物、或其盐、或其立体异构体、或其溶剂合物、或其水合物、或其前药在制备cdk4和/或cdk6激酶抑制剂中的用途。
[0110]
进一步地,所述激酶抑制剂为预防和/或治疗肿瘤的药物;
[0111]
优选地,所述的肿瘤为脑癌、成胶质细胞瘤、白血病、淋巴瘤、bannayan-zonana综合征、考登病、lhermitte-duclos病、乳腺癌、炎性乳腺癌、维尔姆斯瘤、尤因肉瘤、横纹肌肉瘤、室管膜瘤、成神经管细胞瘤、结肠癌、胃癌、膀胱癌、头颈癌、肾癌、肺癌、肝癌、黑素瘤、肾脏癌、卵巢癌、胰腺癌、前列腺癌、肉瘤、骨肉瘤、骨巨细胞瘤或甲状腺癌。
[0112]
进一步地,所述激酶抑制剂为预防和/或治疗自身免疫性疾病的药物;
[0113]
优选地,所述自身免疫性疾病为银屑病或红斑狼疮。
[0114]
本发明还提供了一种药物,它是以前述的化合物、或其盐、或其立体异构体、或其溶剂合物、或其水合物、或其前药为活性成分,加上药物上可接受的辅料或辅助性成分制备而成的药物制剂。
[0115]
本发明中提供的化合物和衍生物根据iupac(国际纯粹与应用化学联合会)或cas(化学文摘服务社,columbus,oh)命名系统命名。
[0116]
本发明中,碳氢基团中碳原子含量的最小值和最大值通过前缀表示,例如,c
a~b
烷基表明任何含“a”至“b”个碳原子的烷基。因此,例如,c1~c8烷基是指包含1~8个碳原子的直链或支链烷基;c1~c8烷氧基是指包含1~8个碳原子的烷氧基。
[0117]
本发明中,卤素为氟、氯、溴或碘。
[0118]
本发明式i中,n1为1时,结构为n1为2时,结构为n1为3时,结构为
[0119]
本发明式i中,虚线为无时,结构为虚线为键时,结构为n2为0、1、2、3、4或5。
[0120]
本发明提供了一种吡啶-嘧啶胺-苯并咪唑衍生物,该衍生物对cdk4、cdk6激酶具有良好的抑制活性,可用于制备cdk4和cdk6激酶抑制剂;同时,该衍生物对多种肿瘤细胞具有显著的抑制活性,可抑制体内肿瘤生长,效果优异;本发明衍生物给药剂量小,安全性高;此外,本发明衍生物可通过血脑屏障,解决了脑肿瘤治疗的最大难题之一。本发明衍生物可用于制备预防和/或治疗肿瘤及自身免疫性疾病药物,具有良好的应用前景。
[0121]
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
[0122]
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说
明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
[0123]
图1为各组nod-bablc小鼠的肿瘤生长情况。
[0124]
图2为化合物a2不同时间点在血液和脑组织中的分布情况。
具体实施方式
[0125]
除另有说明外,本发明具体实施方式中使用的原料、设备均为已知产品,通过购买市售产品获得。
[0126]
实施例1、化合物a1 5-氟-4-(4-氟-1-异丙基-2-甲基-1h-苯并[d]咪唑-6-基)-n-(5-(4-(2-甲氧基乙基)哌嗪-1-基)甲基)吡啶-2-基)嘧啶-2-胺
[0127][0128]
化合物a1的合成路线如下:
[0129][0130]
第一步:
[0131]
将原料1(1g,4.61mmol)溶解于无水dmf(15ml)中,加入无水碳酸钾(862mg,6.24mmol),常温条件下搅拌5min。随后缓慢加入原料2(1.03g,5.532mmol),55℃下搅拌反应2h。tlc监测反应完全,将反应液冷却至室温,搅拌条件下加入适量冰水,反应体系中有大量白色固体析出,过滤,滤饼用水洗涤3次,烘干得中间体3,白色固体1.321g,收率89%。
[0132]
第二步:
[0133]
将中间体3(1.2g,3.72mmol)溶于甲醇(25ml)中,加入10%的钯碳(396mg,0.372mmol),抽真空,用氢气置换三次后于氢气环境常温搅拌反应2h。tlc监测反应完全,反应液用硅藻土抽滤,用二氯甲烷/甲醇(10/1,v/v)混合溶剂洗涤3次,滤液减压浓缩至干,得粗产物中间体4。未经进一步纯化,直接用于下一步反应。
[0134]
第三步:
[0135]
往中间体4(3.72mmol)、原料5(1.282g,3.72mmol)、pd2(dba)3(341mg,0.372mmol)、x-phos(178mg,0.372mmol)和碳酸铯(1.82g,5.58mmol)的混合体系中加入甲苯(20ml),抽真空,氮气置换3次,反应液在氮气环境中100℃下搅拌反应5h。tlc监测反应完全,将反应液冷却至室温,硅藻土抽滤,用二氯甲烷/甲醇(10/1,v/v)混合溶剂洗涤3次,滤液减压浓缩后
经硅胶柱层析分离得中间体6,米白色固体1.1g,收率51%。
[0136]
第四步:
[0137]
将中间体6(1g,1.73mmol)溶于二氯甲烷(15ml)中,搅拌条件下缓慢滴加三氟乙酸(4ml),常温反应2h。tlc监测反应完全,将反应液减压浓缩至干,再次用少量二氯甲烷溶解,旋干,反复操作3次,得粗产物中间体7。未经进一步纯化,直接用于下一步反应。
[0138]
第五步:
[0139]
将中间体7(0.173mmol)溶于无水dmf(4ml)中,加入碳酸铯(113mg,0.346mmol),常温搅拌5min后缓慢加入原料8(25μl,0.2595mmol),升温至60℃反应3h。tlc监测反应完全,将反应液冷却至室温,加入适量冰水,用乙酸乙酯萃取三次,合并有机相,水洗,再用饱和食盐水洗涤,无水硫酸钠干燥后过滤,滤液减压浓缩至干,进一步用硅胶柱层析分离得目标化合物a1,白色固体68mg,收率73%。hr-ms(esi-tof)m/z:calcd for c
28h34
f2n8o[m+h]
+
537.2902,found 537.2900.
[0140]
实施例2、化合物a2 5-氟-4-(4-氟-1-异丙基-2-甲基-1h-苯并[d]咪唑-6-基)-n-(5-(4-(2-(2-甲氧基乙氧基)乙基)哌嗪-1-基)甲基)吡啶-2-基)嘧啶-2-胺
[0141][0142]
以中间体7和1-溴-2-(2-甲氧基乙氧基)乙烷为原料,参照化合物a1第五步合成方法,得化合物a2,白色固体,收率78%。hr-ms(esi-tof)m/z:calcd for c
30h38
f2n8o2[m+h]
+
581.3164,found 581.3163.
[0143]
实施例3、化合物a3 5-氟-4-(4-氟-1-异丙基-2-甲基-1h-苯并[d]咪唑-6-基)-n-(5-(4-(2-(2-甲氧基乙氧基)乙氧基)乙基)哌嗪-1-基)甲基)吡啶-2-基)嘧啶-2-胺
[0144][0145]
以中间体7和二乙二醇-2-溴乙基甲醚为原料,参照化合物a1第五步合成方法,得化合物a3,白色固体,收率69%。hr-ms(esi-tof)m/z:calcd for c
32h42
f2n8o3[m+h]
+
625.3426,found 625.3425.
[0146]
实施例4、化合物a4(5-((5-氟-4-(4-氟-1-异丙基-2-甲基-1h-苯并[d]咪唑-6-基)嘧啶-2-基)氨基)吡啶-2-基)(4-(2-甲氧基乙基)哌嗪-1-基)甲酮
[0147]
[0148]
化合物a4的合成路线如下:
[0149][0150]
第一步:
[0151]
将原料9(2g,11.9mmol)溶于无水thf(30ml)中,依次加入diea(2.95ml,17.85mmol)、hatu(4.98g,13.09mmol),常温搅拌10min后缓慢加入原料2(3.32g,17.85mmol),常温条件继续搅拌反应4h。tlc监测反应完全,加入适量水,用乙酸乙酯萃取三次,合并有机相,水洗,再用饱和食盐水洗涤,无水硫酸钠干燥后过滤,滤液减压浓缩至干,进一步用硅胶柱层析分离得中间体10,白色固体2.092g,收率52%。
[0152]
第二步:
[0153]
以中间体10为原料,参照化合物a1第二步合成方法,得粗产物中间体11。未经进一步纯化,直接用于下一步反应。
[0154]
第三步:
[0155]
以中间体11和原料5为原料,参照化合物a1第三步合成方法,得中间体12,黄色固体,收率82%。
[0156]
第四步:
[0157]
以中间体12为原料,参照化合物a1第四步合成方法,得粗产物中间体13。未经进一步纯化,直接用于下一步反应。
[0158]
第五步:
[0159]
以中间体13和原料8为原料,参照化合物a1第五步合成方法,得化合物a4,黄色固体,收率77%。hr-ms(esi-tof)m/z:calcd for c
28h32
f2n8o2[m+h]
+
551.2694,found 551.2692.
[0160]
实施例5、化合物a5(5-((5-氟-4-(4-氟-1-异丙基-2-甲基-1h-苯并[d]咪唑-6-基)嘧啶-2-基)氨基)吡啶-2-基)(4-(2-(2-甲氧基乙氧基)乙基)哌嗪-1-基)甲酮
[0161][0162]
以中间体13和1-溴-2-(2-甲氧基乙氧基)乙烷为原料,参照化合物a1第五步合成方法,得化合物a5,黄色固体,收率66%。hr-ms(esi-tof)m/z:calcd for c
30h36
f2n8o3[m+h]
+
595.2956,found 595.2952.
[0163]
实施例6、化合物a6(5-((5-氟-4-(4-氟-1-异丙基-2-甲基-1h-苯并[d]咪唑-6-基)嘧啶-2-基)氨基)吡啶-2-基)(4-(2-(2-甲氧基乙氧基)乙氧基)哌嗪-1-基)甲酮
[0164][0165]
以中间体13和二乙二醇-2-溴乙基甲醚为原料,参照化合物a1第五步合成方法,得化合物a6,黄色固体,收率74%。hr-ms(esi-tof)m/z:calcd for c
32h40
f2n8o4[m+h]
+
639.3219,found 639.3218.
[0166]
实施例7、化合物a7 5-氟-4-(4-氟-1-异丙基-2-甲基-1h-苯并[d]咪唑-6-基)-n-(6-((4-(2-甲氧基乙基)哌嗪-1-基)甲基)吡啶-3-基)嘧啶-2-胺
[0167][0168]
化合物a7的合成路线如下:
[0169][0170]
第一步:
[0171]
将中间体11(918mg,3.0mmol)溶于无水thf(20ml)中,缓慢滴加硼烷/二甲硫醚络合物bh3/s(ch3)2(2m in thf)(6ml),随后将反应体系升温至60℃,搅拌反应3h。tlc监测反应完全,将反应液冷却至室温,加入哌啶(2.5ml)继续常温搅拌30min。将反应液减压浓缩至干,进一步用硅胶柱层析分离得中间体14,白色固体362mg,收率41%。
[0172]
第二步:
[0173]
以中间体14和原料5为原料,参照化合物a1第三步合成方法,得中间体15,白色固体,收率76%。
[0174]
第三步:
[0175]
以中间体15为原料,参照化合物a1第四步合成方法,得粗产物中间体16。未经进一步纯化,直接用于下一步反应。
[0176]
第四步:
[0177]
以中间体16和原料8为原料,参照化合物a1第五步合成方法,得化合物a7,白色固体,收率63%。hr-ms(esi-tof)m/z:calcd for c
28h34
f2n8o[m+h]
+
537.2902,found 537.2902.
[0178]
实施例8、化合物a8 5-氟-4-(4-氟-1-异丙基-2-甲基-1h-苯并[d]咪唑-6-基)-n-(6-((4-(2-(2-甲氧基乙氧基)乙基)哌嗪-1-基)甲基)吡啶-3-基)嘧啶-2-胺
[0179][0180]
以中间体16和1-溴-2-(2-甲氧基乙氧基)乙烷为原料,参照化合物a1第五步合成方法,得化合物a8,白色固体,收率70%。hr-ms(esi-tof)m/z:calcd for c
30h38
f2n8o2[m+h]
+
581.3164,found 581.3161.
[0181]
实施例9、化合物a9 5-氟-4-(4-氟-1-异丙基-2-甲基-1h-苯并[d]咪唑-6-基)-n-(6-((4-(2-(2-甲氧基乙氧基)乙氧基)乙基)哌嗪-1-基)甲基)吡啶-3-基)嘧啶-2-胺
[0182][0183]
以中间体16和二乙二醇-2-溴乙基甲醚为原料,参照化合物a1第五步合成方法,得化合物a9,淡黄色固体,收率59%。hr-ms(esi-tof)m/z:calcd for c
32h42
f2n8o3[m+h]
+
625.3426,found 625.3431.
[0184]
实施例10、化合物a10(4-((5-氟-4-(4-氟-1-异丙基-2-甲基-1h-苯并[d]咪唑-6-基)嘧啶-2-基)氨基)-3-甲氧基苯基)(4-(2-(2-甲氧基乙氧基)乙基)哌嗪-1-基)甲酮
[0185][0186]
化合物a10的合成路线如下:
[0187][0188]
第一步:
[0189]
以原料17为原料,参照化合物a4第一步合成方法,得中间体18,白色固体,收率69%。
[0190]
第二步:
[0191]
以中间体18为原料,参照化合物a1第二步合成方法,得粗产物中间体19。未经进一
步纯化,直接用于下一步反应。
[0192]
第三步:
[0193]
以中间体19和原料5为原料,参照化合物a1第三步合成方法,得中间体20,黄色固体,收率77%。
[0194]
第四步:
[0195]
以中间体20为原料,参照化合物a1第四步合成方法,得粗产物中间体21。未经进一步纯化,直接用于下一步反应。
[0196]
第五步:
[0197]
以中间体21和原料8为原料,参照化合物a1第五步合成方法,得化合物a10,黄色固体,收率65%。hr-ms(esi-tof)m/z:calcd for c
32h39
f2n7o4[m+na]
+
646.2930,found 646.2950.
[0198]
实施例11、化合物a11(4-((5-氟-4-(4-氟-1-异丙基-2-甲基-1h-苯并[d]咪唑-6-基)嘧啶-2-基)氨基)-3-甲氧基苯基)(4-(2-(2-甲氧基乙氧基)乙氧基)哌嗪-1-基)甲酮
[0199][0200]
以中间体21和1-溴-2-(2-甲氧基乙氧基)乙烷为原料,参照化合物a1第五步合成方法,得化合物a11,黄色固体,收率71%。hr-ms(esi-tof)m/z:calcd for c
34h43
f2n7o5[m+na]
+
690.3192,found 690.3185.
[0201]
实施例12、化合物a12 5-氟-4-(4-氟-1-异丙基-2-甲基-1h-苯并[d]咪唑-6-基)-n-(5-(4-(2-甲氧基乙基)哌嗪-1-基)吡啶-2-基)嘧啶-2-胺
[0202][0203]
化合物a12的合成路线如下:
[0204][0205]
第一步:
[0206]
将5-溴-2-硝基吡啶(1g,4.93mmol)溶解于无水dmf(15ml)中,加入无水碳酸钾
n-(5-(4-(2-(2-甲氧基乙氧基)乙氧基)乙基)哌嗪-1-基)吡啶-2-基)嘧啶-2-胺
[0219][0220]
以中间体25和二乙二醇-2-溴乙基甲醚为原料,参照化合物a12第五步合成方法,得化合物a14,白色固体,收率70%。hr-ms(esi-tof)m/z:calcd for c
31h40
f2n8o3[m+h]
+
610.3191,found 610.3191.
[0221]
实施例15、化合物a15 5-氟-4-(4-氟-1-异丙基-2-甲基-1h-苯并[d]咪唑-6-基)-n-(6-((4-(2-甲氧基乙基)哌嗪-1-基)吡啶-3-基)嘧啶-2-胺
[0222][0223]
化合物a15的合成路线如下:
[0224][0225]
第一步:
[0226]
将2-溴-5-硝基吡啶(1g,4.93mmol)溶解于无水dmf(15ml)中,加入无水碳酸钾(922mg,6.70mmol),常温条件下搅拌5min。随后缓慢加入原料2(1.02g,5.530mmol),55℃下搅拌反应2h。tlc监测反应完全,将反应液冷却至室温,搅拌条件下加入适量冰水,反应体系中有大量白色固体析出,过滤,滤饼用水洗涤3次,烘干得中间体26,白色固体1.210g,收率89%。
[0227]
第二步:
[0228]
将中间体26(1.19g,3.88mmol)溶于甲醇(25ml)中,加入10%的钯碳(395mg,0.371mmol),抽真空,用氢气置换三次后于氢气环境常温搅拌反应2h。tlc监测反应完全,反应液用硅藻土抽滤,用二氯甲烷/甲醇(10/1,v/v)混合溶剂洗涤3次,滤液减压浓缩至干,得粗产物中间体27。未经进一步纯化,直接用于下一步反应。
[0229]
第三步:
[0230]
以中间体27和原料5为原料,参照化合物a12第三步合成方法,得中间体28,黄色固体,收率78%。
[0231]
第四步:
[0232]
以中间体28为原料,参照化合物a12第四步合成方法,得粗产物中间体29。未经进一步纯化,直接用于下一步反应。
[0233]
第五步:
[0234]
以中间体29和原料8为原料,参照化合物a12第五步合成方法,得化合物a15,白色固体,收率63%。hr-ms(esi-tof)m/z:calcd for c
27h32
f2n8o[m+h]
+
522.2667,found 522.2667.
[0235]
实施例16、化合物a16 5-氟-4-(4-氟-1-异丙基-2-甲基-1h-苯并[d]咪唑-6-基)-n-(6-((4-(2-(2-甲氧基乙氧基)乙基)哌嗪-1-基)吡啶-3-基)嘧啶-2-胺
[0236][0237]
以中间体29和1-溴-2-(2-甲氧基乙氧基)乙烷为原料,参照化合物a12第五步合成方法,得化合物a16,白色固体,收率70%。hr-ms(esi-tof)m/z:calcd for c
29h36
f2n8o2[m+h]
+
566.2929,found 566.2929.
[0238]
实施例17、化合物a17 5-氟-4-(4-氟-1-异丙基-2-甲基-1h-苯并[d]咪唑-6-基)-n-(6-((4-(2-(2-甲氧基乙氧基)乙氧基)乙基)哌嗪-1-基)吡啶-3-基)嘧啶-2-胺
[0239][0240]
以中间体29和二乙二醇-2-溴乙基甲醚为原料,参照化合物a1第五步合成方法,得化合物a17,淡黄色固体,收率59%。hr-ms(esi-tof)m/z:calcd for c
31h40
f2n8o3[m+h]
+
610.3191,found 610.3191.
[0241]
实施例18、化合物a18 5-氟-4-(4-氟-1-异丙基-2-甲基-1h-苯并[d]咪唑-6-基)-n-(5-(4-(2-甲氧基乙基)哌嗪-1-基)苯基-2-基)嘧啶-2-胺
[0242][0243]
化合物a18的合成路线如下:
[0244][0245]
第一步:
[0246]
将1-溴-4-硝基苯(1g,4.61mmol)溶解于无水dmf(15ml)中,加入无水碳酸钾(860mg,6.22mmol),常温条件下搅拌5min。随后缓慢加入原料2(1.01g,5.530mmol),55℃下搅拌反应2h。tlc监测反应完全,将反应液冷却至室温,搅拌条件下加入适量冰水,反应体系中有大量白色固体析出,过滤,滤饼用水洗涤3次,烘干得中间体30,白色固体1.221g,收率86%。
[0247]
第二步:
[0248]
将中间体30(1.2g,3.71mmol)溶于甲醇(25ml)中,加入10%的钯碳(395mg,0.371mmol),抽真空,用氢气置换三次后于氢气环境常温搅拌反应2h。tlc监测反应完全,反应液用硅藻土抽滤,用二氯甲烷/甲醇(10/1,v/v)混合溶剂洗涤3次,滤液减压浓缩至干,得粗产物中间体31。未经进一步纯化,直用于接下一步反应。
[0249]
第三步:
[0250]
往中间体31(3.72mmol)、原料5(1.281g,3.71mmol)、pd2(dba)3(340mg,0.3712mmol)、x-phos(177mg,0.371mmol)和碳酸铯(1.81g,5.57mmol)的混合体系中加入甲苯(20ml),抽真空,氮气置换3次,反应液在氮气环境中100℃下搅拌反应5h。tlc监测反应完全,将反应液冷却至室温,硅藻土抽滤,用二氯甲烷/甲醇(10/1,v/v)混合溶剂洗涤3次,滤液减压浓缩后经硅胶柱层析分离得中间体32,白色固体1.05g,收率50%。
[0251]
第四步:
[0252]
将中间体32(1.0g,1.73mmol)溶于二氯甲烷(15ml)中,搅拌条件下缓慢滴加三氟乙酸(4ml),常温反应2h。tlc监测反应完全,将反应液减压浓缩至干,再次用少量二氯甲烷溶解,旋干,反复操作3次,得粗产物中间体33。未经进一步纯化,直接用于下一步反应。
[0253]
第五步:
[0254]
将中间体33(0.173mmol)溶于无水dmf(4ml)中,加入碳酸铯(113mg,0.346mmol),常温搅拌5min后缓慢加入原料8(25μl,0.2595mmol),升温至60℃反应3h。tlc监测反应完全,将反应液冷却至室温,加入适量冰水,用乙酸乙酯萃取三次,合并有机相,水洗,再用饱和食盐水洗涤,无水硫酸钠干燥后过滤,滤液减压浓缩至干,进一步用硅胶柱层析分离得目标化合物a18,白色固体68mg,收率73%。hr-ms(esi-tof)m/z:calcd for c
28h33
f2n7o[m+h]
+
522.2715,found 522.2715.
[0255]
实施例19、化合物a19 5-氟-4-(4-氟-1-异丙基-2-甲基-1h-苯并[d]咪唑-6-基)-n-(5-(4-(2-(2-甲氧基乙氧基)乙基)哌嗪-1-基)苯基-2-基)嘧啶-2-胺
[0256][0257]
以中间体33和1-溴-2-(2-甲氧基乙氧基)乙烷为原料,参照化合物a1第五步合成方法,得化合物a2,白色固体,收率78%。hr-ms(esi-tof)m/z:calcd for c
30h37
f2n7o2[m+h]
+
565.2977,found 565.2977.
[0258]
实施例20、化合物a20 5-氟-4-(4-氟-1-异丙基-2-甲基-1h-苯并[d]咪唑-6-基)-n-(5-(4-(2-(2-甲氧基乙氧基)乙氧基)乙基)哌嗪-1-基)苯基-2-基)嘧啶-2-胺
[0259][0260]
以中间体33和二乙二醇-2-溴乙基甲醚为原料,参照化合物a1第五步合成方法,得化合物a20,白色固体,收率68%。hr-ms(esi-tof)m/z:calcd for c
32h41
f2n7o3[m+h]
+
609.3239,found 609.3239.
[0261]
通过以下体外试验证明本发明的有益效果。
[0262]
试验例1、cdk4、cdk6激酶抑制活性测试
[0263]
本实验的目的是检测发明化合物对体外激酶的抑制活性,采用的方法为同位素标记法。本实验分别对cdk4、cdk6激酶进行体外活性抑制测试。abemaciclib为阳性对照。受试化合物的激酶抑制活性用ic
50
(半数抑制浓度)。ic
50
值可通过受试化合物在一系列不同浓度下对激酶活性的抑制率计算而获得。
[0264]
1、实验材料
[0265]
20mm 3-(n-吗啉基)丙磺酸(mops);1mm乙二胺四乙酸(edta);0.01%布里杰35(brij-35);5%甘油(glycerol);0.1%巯基乙醇(mercptoethanol);1mg/ml的牛血清白蛋白(bsa);l0mm的二氯化锰溶液(mnc12);终止缓冲液(3%磷酸盐缓冲液);洗涤缓冲液(75mm的磷酸盐溶液);甲醇(methanol);filtermat a膜;cdk4/6激酶,受试化合物。
[0266]
2、实验方法
[0267]
将待测的激酶及相应的底物与不同浓度的待测化合物或dmso一同加入到反应缓冲液(8mm丙磺酸盐,0.2mm edta,10mm醋酸镁和km浓度的γ-33p-atp溶液)中,室温孵育40min;加入3%的磷酸盐溶液以终止反应;吸取10μl反应混合液滴在p30滤纸上;用75mm的磷酸盐溶液清洗滤纸3次;再用甲醇清洗滤纸1次;滤纸晾干后加入闪烁液,通过闪烁计数测量磷酸化的底物。抑制率%=(1-化合物处理组计数值/空白对照组计数值)
×
100%;半数抑制浓度(ic
50
)由各浓度对应的抑制率拟合得到。
[0268]
3、实验结果
[0269]
通过以上实验方法,测试了本发明中的化合物分别针对cdk4/6激酶的抑制活性。表1给出了受试化合物对cdk4/6激酶抑制活性的ic
50
值。
[0270]
实验结果表明,受试化合物对cdk4/6激酶具有很强的抑制活性。可以用于制备cdk4激酶和cdk6激酶抑制剂。
[0271]
表1.受试化合物对cdk4/6激酶抑制活性的ic
50
值
[0272][0273]
*a:ic
50
《50nm;b:50nm≤ic
50
《100nm;c:100nm≤ic
50
《150nm;d:ic
50
>150nm
[0274]
试验例2、化合物的体外肿瘤细胞增殖抑制实验
[0275]
本实验的目的是检测发明化合物对体外肿瘤细胞增殖抑制活性,采用的方法为mtt(四甲基偶氮唑盐)比色法。
[0276]
1、实验材料
[0277]
1.1主要试剂
[0278]
乳腺癌细胞株mcf-7、mda-mb-231、mda-mb-436和mda-mb-468,结直肠癌细胞株sw620,肺癌细胞a549均购自于美国标准生物品收藏中心(american type culture collection,atcc),rpmi 1640培养基,胎牛血清(fbs)均购买自美国gibico公司;青霉素和链霉素购自大连宝生物公司;培养细胞用平皿和96孔板等购买于corning公司;各种规格的离心管购自bd公司;mtt试剂购自日本同仁化学研究所(donjindo)。受试化合物由发明人合成,体外实验时用100%dmso配制成10mm储存液,置-20℃冰箱避光保存备用,测试时用培养液稀释至所需浓度。
[0279]
1.2细胞系及培养
[0280]
本实验所用乳腺癌细胞株mcf-7、mda-mb-231、mda-mb-436和mda-mb-468,结直肠癌细胞株sw620,肺癌细胞a549用含10%胎牛血清(fbs)、100iu/ml青霉素、100μg/ml链霉素的常规rpmi 1640完全培养基培养于37℃、5%co2的培养箱中培养。
[0281]
2、实验方法
[0282]
用完全细胞培养液调整细胞浓度为1~2
×
104个/ml的细胞悬液(hcc827细胞浓度
为6
×
104个/ml,h1975细胞为4
×
103个/ml),接种于96孔板,每孔200μl细胞悬液,培养过夜。次日,吸弃上清,然后分别用梯度浓度的受试化合物处理细胞。同时设不含药物的阴性对照组和等体积的溶剂对照组,dmso浓度为0.1%,每个剂量组设3个复孔,在37℃,5%co2条件下培养。72小时后,每孔加入浓度为5mg/ml的mtt试剂20μl,再培养2-4h后,弃上清,每孔再加入dmso 150μl,振荡混匀15min,用酶标仪(λ=570nm)测定吸光度(a)值(a值与活细胞数成正比),取其平均值。相对细胞增殖抑制率=(对照组a570-实验组a570)/对照组a570
×
100%。实验至少重复3次。实验数据用均数表示,数据统计资料采用t检验,p《0.05为差异有统计学意义。以下各化合物对细胞增殖抑制作用均用ic
50
表示。
[0283]
3、实验结果
[0284]
采用以上方法,对乳腺癌细胞株mcf-7、mda-mb-231、mda-mb-436和mda-mb-468,结直肠癌细胞株sw620,肺癌细胞a549进行了增殖抑制活性测试。表2给出了受试化合物增殖抑制活性(ic
50
)。结果表明,大部分实施例化合物对所测试肿瘤细胞株均展现出显著的增殖抑制活性,其中化合物a8和a9对多种肿瘤细胞株的增殖抑制活性均在低微摩尔级,相比于阳性化合物abemaciclib具有一定治疗优势。
[0285]
表2.化合物对肿瘤细胞株的增殖作用(mtt法)
[0286][0287]
注:nd:not determined.
[0288]
a:ic
50
《0.1μm;b:0.1μm≤ic
50
《1μm;c:ic
50
≥1μm.
[0289]
试验例3、化合物a2的体内抗肿瘤实验
[0290]
本实验的目的是检测发明化合物的体内抗肿瘤效果。实验用balb/c小鼠皮下瘤模型,测试化合物a2的体内抗肿瘤活性。所用的细胞株为人结肠癌细胞株colo205。
[0291]
1、实验材料
[0292]
胎牛血清、培养基、胰酶等购自gibco brl公司(invitrogen corporation,usa),培养基购自atcc(american type culture collection),人结肠癌细胞株colo205购自美
国atcc公司,nod-bablc小鼠购自于北京华阜康动物实验中心。
[0293]
2、实验方法
[0294]
使用6-8周的nod-bablc小鼠,按照约1
×
107个/0.1ml/只colo205细胞浓度接种于小鼠皮下后肋部,带肿瘤长到一定体积后,小鼠随机分组并开始灌胃口服及尾静脉给药。
[0295]
观察指标:每3天测量一次小鼠体重计肿瘤长径、短颈并计算肿瘤体积(长径
×
短径2
×
0.52),观察有无腹泻、抽筋、皮疹,体重明显降低等反应。
[0296]
3、实验结果
[0297]
实验测得的各种不同组别的肿瘤生长情况如图1所示。实验结果说明,受试化合物a2对colo205具有明显的体内生长抑制作用。在给药的过程中未发现小鼠出现体重降低、皮疹、腹泻等不良反应,表明在测试剂量下,受试化合物a2在给药剂量范围内毒性很低。
[0298]
试验例4、化合物a2的血脑组织分布实验
[0299]
本实验的目的是检测发明化合物的体内抗肿瘤效果。实验用balb/c小鼠脑肿瘤原位模型,测试化合物a2的体内抗肿瘤活性。所用的细胞株为人脑星形胶质母细胞u87-luc。
[0300]
1、实验材料
[0301]
胎牛血清、培养基、胰酶等购自gibco brl公司(invitrogen corporation,usa),培养基购自atcc(american type culture collection),nod-bablc小鼠购自于北京华阜康动物实验中心。
[0302]
2、实验方法
[0303]
使用spf级icr小鼠,给药前称重,根据体重,计算给药量。通过静脉注射给药。
[0304]
血浆样本处理:采集经心脏穿刺采血,每个样品采集约0.2ml,肝素钠抗凝,采集后放置冰上,并于1小时之内离心分离血浆(离心条件:6800g,6分钟,2-8℃)。血浆样本在分析前存放时则放于-80℃冰箱内。
[0305]
组织样品处理:动物安乐死后,采集脑组织,用生理盐水冲洗,避免交叉污染,滤纸吸干,然后放入标记好的管子/自封袋中(一个组织一个管子/自封袋),样品在存放入超低温冰箱前临时放置在冰上。
[0306]
实验分组:药物溶剂对照组(12.5%el+12.5%etoh+75%水)
[0307]
化合物a2-m:50mg/kg q.d;
[0308]
化合物a2-h:100mg/kg q.d;
[0309]
化合物abemaciclib:50mg/kg q.d;
[0310]
(各组药物溶解于12.5%el+12.5%etoh+75%水)
[0311]
结果分析:通过不同时间点的血药浓度数据,运用phoenix winnonlin7.0计算药代动力学参数,提供auc0-t、auc0-∞、mrt0-∞、cmax、tmax、和t1/2等参数及其平均值和标准差。
[0312]
3、实验结果
[0313]
化合物a2不同时间点在血液和脑组织中的分布情况如图2所示。实验结果表明,受试化合物a2在不同时间点时,在脑组织中的药物浓度均高于其在血浆中的浓度。说明化合物a2能够透过血脑屏障,直接进入脑部发挥作用,可用于针对脑肿瘤药物的开发。
[0314]
综上,本发明提供了一种吡啶-嘧啶胺-苯并咪唑衍生物,该衍生物对cdk4、cdk6激酶具有良好的抑制活性,可用于制备cdk4和cdk6激酶抑制剂;同时,该衍生物对多种肿瘤细
胞具有显著的抑制活性,可抑制体内肿瘤生长,效果优异;本发明衍生物给药剂量小,安全性高;此外,本发明衍生物可通过血脑屏障,解决了脑肿瘤治疗的最大难题之一。本发明衍生物可用于制备预防和/或治疗肿瘤及自身免疫性疾病药物,具有良好的应用前景。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1