一种肟醚的制备方法
1.本发明属于杀菌剂、农药及医药合成技术领域,具体涉及一种肟醚的制备方法。
背景技术:
2.肟醚及其衍生物是一种重要的化工产品,在农药和医药上有很重要的用途。因其具有高效、低毒及低残留等优点,而常被用作制备重要新型药物及抗菌剂常用的有效基团。目前关于肟醚的合成方法已经有些报道。如在cn109369449a中报道了将酮肟与有机碱按比例投入高压反应釜中,控制反应温度在10-60℃之间,并通入烷基化试剂反应2-6h,反应结束后进行过滤,滤液脱溶后结晶可得到肟醚,产率约为85%。在欧洲专利ep0158159中采用了水相分步法工艺,即先将酮肟与氢氧化钠中反应得到肟钠盐,再加入烷基化试剂进行烷基化反应,反应完成后进行萃取,精馏得到肟醚产品。上述工艺路线相对复杂,操作较繁琐,生产成本高,后处理相对复杂。2016年,任志辉课题组开发了以肟与四氢呋喃为原料,通过cui催化发生脱氢交叉偶联反应,实现构建c-o键合成肟醚。此反应虽然工艺流程简单,但使用了金属催化剂,在进行废液处理时容易有金属残留,对环境以及人体都存在一定的危害。因此,市场亟需一种反应体系简单,反应温和,成本低廉的肟醚合成工艺。
技术实现要素:
3.针对现有技术中的问题,本发明解决了现有工艺的难点,提供了一种安全、绿色、操作简单的肟醚制备方法,利用可见光介导的无金属催化构建c-o键合成肟醚。
4.为实现以上技术目的,本发明的技术方案是:
5.一种肟醚的制备方法,所述肟醚的结构如式(1)所示:
[0006][0007]
其中,所述r1采用p-ph、p-cl、p-f、p-c(ch3)3、p-ome、p-ch3、p-cf3、o,p-(ch3)2、o,m,p-f5、syn-(ch3)3、ph中的一种;
[0008]
所述r2采用ch3、ch2ch3、ph-ch3、h、环氧己烷、4-cl-ph、4-f-ph、4-me-ph、ph、(ch2)2ch3中的一种;
[0009]
所述r采用所述r采用中的一种。
[0010]
进一步的,所述肟醚的制备方法是以含有r1、r2基团的酮肟或醛肟为原料,加入烷基化试剂和除水剂,以四溴化碳为催化剂,在有机溶剂和可见光照射条件下通过四溴化碳产生的溴自由基催化夺取烷基化试剂的氢,产生的自由基进行单电子转移以及与肟进行亲
电取代反应得到肟醚产物。
[0011]
所述原料采用对r1、r2基团的酮肟或醛肟,进一步的,所述原料采用苯乙酮肟、对氯苯乙酮肟、对三氟苯甲醛肟、二苯甲酮肟、2,4-二甲基苯甲醛肟、对甲基苯甲醛肟、苯丙酮肟中的一种。
[0012]
所述烷基化试剂为环氧己烷、四氢呋喃、2-甲基四氢呋喃、四氢吡喃中的一种。
[0013]
所述有机溶剂为为乙酸正丁酯、乙酸乙酯、四氢呋喃、乙醇、甲苯中的一种。
[0014]
所述反应时间为4-6h。
[0015]
所述四溴化碳的用量为0-2.0eq。
[0016]
所述除水剂采用无水硫酸镁、4a分子筛中的一种。
[0017]
所述肟醚产物的提纯方法是在反应结束后,以石油醚和乙酸乙酯为展开剂,用柱层析进行分离,脱溶浓缩后进行干燥。
[0018]
四溴化碳作为一种廉价易合成的有机小分子试剂,在加热或者光照的条件下会使c-br键发生同源裂解产生高活性的溴自由基。它通常具有很好的化学是选择性和区域选择性,对交叉脱氢偶联反应(cdc)表现出很好的反应活性。
[0019]
从以上描述可以看出,本发明具备以下优点:
[0020]
1.本发明解决了现有工艺的难点,提供了一种安全、绿色、操作简单的肟醚制备方法,利用可见光介导的无金属催化构建c-o键合成肟醚。
[0021]
2.本发明在可见光条件下,通过四溴化碳催化肟与烷基化试剂进行交叉脱氢偶联反应合成肟醚,利用四溴化碳替代金属的复杂催化体系,具有成本低,反应条件温和,时间短,易于处理等优点。
[0022]
3.本发明利用可见光在常温下进行,反应条件温和,避免了高温反应带来的安全隐患,易于控制反应,同时,反应操作过程和后处理简单。
[0023]
4.本发明提供的工艺绿色环保,更适合用于工业化生产,降低制备成本的同时得到高产率、高纯度的产物,表现出更佳的生态优势及经济优势。
附图说明
[0024]
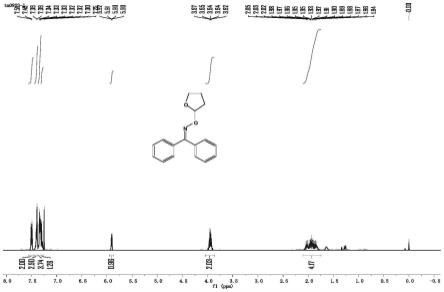
图1为实例1制备的肟醚的核磁氢谱图。
[0025]
图2为实例1制备的肟醚的核磁碳谱图。
[0026]
图3为实例2制备的肟醚的核磁氢谱图。
[0027]
图4为实例2制备的肟醚的核磁碳谱图。
[0028]
图5为实例3制备的肟醚的核磁氢谱图。
[0029]
图6为实例3制备的肟醚的核磁碳谱图。
[0030]
图7为实例4制备的肟醚的核磁氢谱图。
[0031]
图8为实例4制备的肟醚的核磁碳谱图。
[0032]
图9为实例5制备的肟醚的核磁氢谱图。
[0033]
图10为实例5制备的肟醚的核磁碳谱图。
[0034]
图11为实例6制备的肟醚的核磁氢谱图。
[0035]
图12为实例6制备的肟醚的核磁碳谱图。
[0036]
图13为实例7制备的肟醚的核磁氢谱图。
[0037]
图14为实例7制备的肟醚的核磁碳谱图。
[0038]
图15为实例8制备的肟醚的核磁氢谱图。
[0039]
图16为实例8制备的肟醚的核磁碳谱图。
[0040]
图17为实例9制备的肟醚的核磁氢谱图。
[0041]
图18为实例9制备的肟醚的核磁碳谱图。
[0042]
图19为实例10制备的肟醚的核磁氢谱图。
[0043]
图20为实例10制备的肟醚的核磁碳谱图。
具体实施方式
[0044]
结合图1至图20,详细说明本发明的具体实施例,但不对本发明的权利要求做任何限定。
[0045]
实施例1
[0046]
在5ml的透明玻璃反应瓶中依次加入无水乙酸正丁酯、二苯甲酮肟(0.0197g,0.1mmol,1.0eq),无水四氢呋喃(0.3606g,50eq)、四溴化碳(0.0597g,1.8eq)、0.1000g分子筛,混合均匀。放入磁子后将反应瓶置于氩气条件下通气15分钟,在3w led蓝灯的照射下反应5小时。反应完成后,将反应后的溶液置于离心管中离心,取上层澄清液体用旋蒸仪减压蒸馏浓缩得到浓稠液体。最后用乙酸乙酯和石油醚按比例混合作为展开剂,进行柱层析分离,得到的产物脱溶干燥后,产率为87%。如图1所示:1h nmr(400mhz,cdcl3)δ7.56
–
7.46(m,2h),7.44
–
7.37(m,3h),7.37
–
7.31(m,4h),7.30(s,1h),5.91(dd,j=5.3,1.3hz,1h),4.05
–
3.87(m,2h),2.11
–
1.75(m,4h)。如图2所示:13c nmr(101mhz,cdcl3)δ158.15(s),136.49(s),133.55(s),129.35(d,j=7.4hz),128.68(s),128.16(d,j=7.2hz),127.91(s),106.70(s),77.38(s),77.06(s),76.74(s),67.98(s),30.71(s),23.88(s).
[0047]
替换例1-12的制备方法同实施例1,区别在于调整反应时间、有机溶剂种类、四溴化碳的用量等,并分别测试其对反应的影响。
[0048]
[0049][0050]
如上表所述,在肟醚合成过程中,当反应时间为5h,四溴化碳的用量为原料的1.8eq,加入4a分子筛,以乙酸丁酯为溶剂时,得到的肟醚产率最高,产率为87%。
[0051]
实施例2
[0052]
将0.0189g对三氟甲基苯甲醛肟(0.1mmol)代替0.0197g二苯甲酮肟(0.1mmol),其他条件和操作过程同实施例1,得到的纯产品对三氟甲基苯甲醛-o-2-四氢呋喃基肟醚,产率为90%。如图3所示:1h nmr(400mhz,cdcl3)δ8.11(s,1h),7.72(d,j=8.2hz,2h),7.61(d,j=8.3hz,2h),5.90(dd,j=5.0,1.5hz,1h),4.07
–
3.93(m,2h),2.18
–
2.02(m,3h),1.93(dt,j=9.5,4.6hz,1h)。如图4所示:
13
c nmr(101mhz,cdcl3)δ148.54(s),135.59(s),125.57(dd,j=7.5,3.7hz),107.00(s),77.36(s),77.04(s),76.73(s),68.14(s),30.82(s),23.77(s)。
[0053]
实施例3
[0054]
将0.0149g 2,4-二甲基苯甲醛肟(0.1mmol)代替0.0197g二苯甲酮肟(0.1mmol),其他条件和操作过程同实施例1,得到的纯产品为2,4-二甲基苯甲醛-o-2-四氢呋喃基肟醚,产率为93%。如图5所示:1h nmr(400mhz,cdcl3)δ8.32(s,1h),7.64(d,j=7.8hz,1h),6.99(d,j=9.6hz,2h),5.92
–
5.86(m,1h),4.08
–
3.91(m,2h),2.38(s,3h),2.31(s,3h),2.16
–
2.00(m,3h),1.97
–
1.86(m,1h)。如图6所示:
13
c nmr(101mhz,cdcl3)δ148.92(s),139.82(s),136.75(s),131.47(s),127.52(s),127.11(s),127.00(d,j=20.8hz),106.58(s),77.39(s),77.07(s),76.75(s),68.00(s),30.87(s),23.89(s),21.31(s),19.73(s)。
[0055]
实施例4
[0056]
将0.0169g对氯苯乙酮肟(0.1mmol)代替0.0197g二苯甲酮肟(0.1mmol),其他条件和操作过程同实施例1,得到的纯产品为对氯苯乙酮-o-2-四氢呋喃基肟醚,产率为91%。如图7所示:1h nmr(400mhz,cdcl3)δ7.61(d,j=7.3hz,2h),7.31(d,j=7.3hz,2h),5.89(d,j=2.0hz,1h),4.07
–
3.86(m,2h),2.20(s,3h),2.15
–
2.00(m,3h),1.97
–
1.84(m,1h)。如图8所示:
13
c nmr(101mhz,cdcl3)δ154.68(s),135.04(d,j=16.1hz),128.47(s),127.52(s),106.62(s),77.42(s),77.11(s),76.79(s),67.98(s),30.90(s),23.95(s),12.75(s)。
[0057]
实施例5
[0058]
将0.0135g对甲基苯甲醛肟(0.1mmol)代替0.0197g二苯甲酮肟(0.1mmol),其他条件和操作过程同实施例1,得到的纯产品为对甲基苯甲醛-o-2-四氢呋喃基肟醚,产率为88%。如图9所示:1h nmr(400mhz,cdcl3)δ8.05(s,1h),7.50(d,j=8.1hz,2h),7.16(d,j=8.0hz,2h),5.90
–
5.84(m,1h),4.12
–
3.89(m,2h),2.35(s,3h),2.15
–
2.01(m,3h),1.91(dd,j=7.5,2.6hz,1h).如图10所示:
13
c nmr(101mhz,cdcl3)δ150.08(s),140.16(s),129.33(s),127.27(s),106.56(s),77.38(s),77.06(s),76.74(s),67.91(d,j=9.7hz),30.83(s),23.86(s),21.47(s).
[0059]
实施例6
[0060]
将0.0135g苯乙酮肟(0.1mmol)代替0.0197g二苯甲酮肟(0.1mmol),其他条件和操作过程同实施例1,得到的纯产品为苯乙酮-o-2-四氢呋喃基肟醚,产率为93%。如图11所示:1h nmr(400mhz,cdcl3)δ7.68(dd,j=6.7,3.0hz,2h),7.37
–
7.33(m,3h),6.08
–
5.81(m,1h),3.99(ddd,j=13.5,11.5,6.8hz,2h),2.24(s,3h),2.15
–
2.03(m,3h),1.96
–
1.85(m,1h).如图12所示:
13
c nmr(101mhz,cdcl3)δ155.86(s),136.56(s),129.14(s),128.29(s),126.27(s),106.51(s),77.36(s),77.05(s),76.73(s),67.96(s),30.94(s),23.97(s),12.97(s).
[0061]
实施例7
[0062]
将0.0149g苯丙酮肟(0.1mmol)代替0.0197g二苯甲酮肟(0.1mmol),其他条件和操作过程同实施例1,得到的纯产品为苯丙酮-o-2-四氢呋喃基肟醚,产率为83%。如图13所示:1h nmr(400mhz,cdcl3)δ7.67(dd,j=6.8,3.0hz,2h),7.35(dd,j=5.0,1.8hz,3h),5.97
–
5.80(m,1h),3.99(ddd,j=13.4,11.6,6.8hz,2h),2.75(qd,j=7.6,3.3hz,2h),2.18
–
2.01(m,3h),1.93(d,j=1.2hz,1h),1.14(t,j=7.6hz,3h).如图14所示:
13
c nmr(101mhz,cdcl3)δ160.84(s),135.55(s),129.08(s),128.34(s),126.47(s),106.43(s),77.36(s),77.04(s),76.73(s),67.87(s),30.91(s),23.92(s),20.44(s),11.18(s).
[0063]
实施例8
[0064]
将0.0135g苯乙酮肟(0.1mmol)代替0.0197g二苯甲酮肟(0.1mmol),0.1503g环氧己烷(15eq)代替0.3606g四氢呋喃(50eq),其他条件和操作过程同实施例1,得到苯乙酮-o-2-环氧己烷基肟醚,产率为50%。如图15所示:1h nmr(400mhz,cdcl3)δ7.68(dd,j=6.5,2.9hz,2h),7.37
–
7.32(m,3h),5.54(dd,j=9.2,5.2hz,1h),3.94
–
3.84(m,1h),3.71
–
3.61(m,1h),2.27(s,3h),1.91
–
1.81(m,2h),1.81
–
1.35(m,6h).如图16所示:
13
c nmr(101mhz,cdcl3)δ155.96(s),136.55(s),129.19(s),128.32(s),126.32(s),104.60(s),77.40(s),77.08(s),76.77(s),62.82(s),32.89(s),30.77(s),29.42(s),22.92(s),13.12(s).
[0065]
实施例9
[0066]
将0.0135g苯乙酮肟(0.1mmol)代替0.0197g二苯甲酮肟(0.1mmol),0.4307g(50eq)四氢吡喃代替0.3606g四氢呋喃(50eq),其他条件和操作过程同实施例1,得到苯乙酮-o-2-四氢吡喃基肟醚,产率为30%。如图17所示::1h nmr(400mhz,cdcl3)δ7.70
–
7.66(m,2h),7.37
–
7.33(m,3h),5.41(dd,j=4.9,2.6hz,1h),3.94(ddd,j=10.8,7.7,3.0hz,1h),3.68
–
3.60(m,1h),2.31(s,3h),1.94
–
1.76(m,3h),1.62(dd,j=8.7,3.3hz,3h)。如图18所示:
13
c nmr(101mhz,cdcl3)δ156.37(s),136.48(s),129.23(s),128.34(s),126.38(s),101.08(s),77.43(s),77.11(s),76.79(s),29.17(s),25.33(s),20.09(s),13.09(s)。
[0067]
实施例10
[0068]
将0.0135g苯乙酮肟(0.1mmol)代替0.0197g二苯甲酮肟(0.1mmol),0.4301g 2-甲基四氢呋喃(50eq)代替0.3606g四氢呋喃(50eq),其他条件和操作过程同实施例1,得到苯乙酮-o-2-(2-四氢呋喃)基肟醚,产率为37%。如图19所示:1h nmr(400mhz,cdcl3)δ7.68(dd,j=6.8,3.0hz,2h),7.35(dd,j=5.0,1.8hz,3h),5.85(d,j=4.4hz,1h),4.27(dd,j=10.4,4.2hz,1h),2.24(s,3h),2.21
–
2.04(m,3h),1.79
–
1.67(m,1h),1.34(d,j=6.2hz,3h).如图20所示:13c nmr(101mhz,cdcl3)δ155.35(s),136.66(s),129.08(s),128.29(s),126.26(s),106.77(s),77.38(s),77.07(s),76.75(s),32.31(s),31.38(s),22.71(s),13.03(s).
[0069]
实施例11:实施例1的放大反应
[0070]
将1.000g二苯甲酮肟(0.005mol),3.027g四溴化碳(1.8eq),18.283g四氢呋喃(50eq),1.000g 4a分子筛,依次加入到100ml的单口烧瓶中,再加入50ml的无水乙酸正丁酯,混合均匀。加入磁子后,将烧瓶置于氩气的条件下通气30分钟,在30w蓝灯的照射下反应12小时。反应完成后的后处理操作过程同实施例1,得到的肟产率为85%。
[0071]
本发明以酮肟或醛肟为原料,通过光介导四溴化碳催化合成肟醚,反应条件绿色
温和,避免使用过渡金属催化剂,反应时间短,操作过程简单,可以有效替代传统的肟醚合成方法。当以二苯甲酮肟和四氢呋喃为基础时,最佳的条件:反应时间为5h,四溴化碳的用量为原料的1.8eq,加入4a分子筛,以乙酸丁酯为溶剂时,得到的肟醚产率最高。并且在该条件下进行底物适应性拓展,从实施例2-5中可以看出,底物的适应性良好。且通过放大反应之后,产率可以达到85%,非常适合工业化生产。
[0072]
在本技术方案中,肟醚产率可以达到93%,高于文献中以金属催化剂cui催化合成肟醚的最高产率(88%),且用环氧己烷与肟进行反应,得到了苯乙酮-o-2-四氢吡喃基肟醚产率为50%,能够实现cui催化体系完成不了的反应。由此可见可见光介导四溴化碳催化合成肟醚的方法可以达到良好的产率,底物的适应性广泛,且没有金属残留,在制药领域有更广阔的应用前景。
[0073]
可以理解的是,以上关于本发明的具体描述,仅用于说明本发明而并非受限于本发明实施例所描述的技术方案。本领域的普通技术人员应当理解,仍然可以对本发明进行修改或等同替换,以达到相同的技术效果;只要满足使用需要,都在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1