一种苯并二氮卓并吡咯类化合物及其制备方法和应用
1.本发明属于生物技术领域,具体涉及一种苯并二氮卓并吡咯类化合物及其制备方法和应用。该类化合物可用于受体配体结合动力学的研究。
背景技术:
2.精氨酸加压素受体(avpr)是一类g蛋白偶联受体(g protein-coupled receptor,gpcr),主要分为精氨酸加压素v
1a
受体(v
1a
r)、精氨酸加压素v
1b
受体(v
1b
r)和精氨酸加压素v2受体(v2r)三种亚型(n engl j med.2015,372,2207-16.)。其中,v2r集中表达于肾小管的集合管以及髓袢升支粗段,在调节水与氯化钠的重吸收、维持机体水盐平衡和调节血浆渗透压中发挥重要作用(the lancet.2008,371,1624-32.),并通过gs蛋白介导的camp通路参与常染色体显性多囊肾病(adpkd)的进展(j am soc nephrol.2008,19,102-108.)。
3.近期,结合动力学在先导化合物筛选中的应用逐渐受到关注。研究提示,药物药效的发挥取决于药物与靶点的结合,当药物分子与其药物靶点结合形成二元复合物时,才能发挥药效(nature reviews drug discovery.2016,15,87-95.)。结合动力学研究的是药物与其靶点结合与解离的动态过程,主要从动力学角度来探讨药物与其靶点的相互作用(chemmedchem.2015,10,1793-96.)。目前,在传统药物研发中,常以亲和力(affinity)作为筛选先导化合物的指标,但是基于传统评价体系中亲和力等评价指标筛选出的先导化合物,尽管会表现出较高的亲和力,但在后期的药效学评价中,可能会产生较低功效。结合动力学参数停留时间(rt)关注于药物与受体结合形成药物-受体二元复合物的存在时间,决定了药物药效的持续时间(chem rev.2017,117,38-66.)。
4.光控药理学是一种新兴的药物发现策略,它利用光控配体来调节生物活性,具有较高的时空分辨率(chem rev.2018,118,10710-47.)。偶氮键由于其双稳态光化学性质和合成便利性,引入偶氮键是制备光控配体分子的常用方法。偶氮化策略是将偶氮苯部分插入具有生物活性的化合物中,使其在近紫外光照射下的构型从反式转变为顺式。在长波长的光照射或热弛豫下,这种构型变化可以恢复。这种光致异构化引起了几何形状、极性的改变,从而改变了分子的生物活性(j med chem.2020,63,11436-47.)。
5.目前,大多数光控配体的研究是基于光照前后受体亲和力或效能指标的变化,而忽略了受体与配体的结合动力学性质,而这一性质已被证明是发挥药效的必要条件(nature reviews chemistry.2022,6,51-69.)。因此,借助偶氮苯光控元件开发一种可逆的光控开关用于研究配体与受体的结合动力学是有应用意义的。
技术实现要素:
6.本发明的目的是在现有技术的基础上,提供一种苯并二氮卓并吡咯类化合物,作为精氨酸加压素受体光控配体化合物,包括探索取代基大小与位置、特定光照波长等,调节配体构型,进而调控精氨酸加压素受体v2r的抑制状态,用于受体配体结合动力学的研究。
7.本发明的另一目的是提供一种上述苯并二氮卓并吡咯类化合物的制备方法。
8.本发明的第三个目的是提供一种上述苯并二氮卓并吡咯类化合物在制备精氨酸加压素受体拮抗剂药物方面的应用,特别是在制备与精氨酸加压素v2受体拮抗剂结合动力学药理工具相关药物方面的应用。
9.本发明的技术方案如下:
10.式i所示的苯并二氮卓并吡咯类化合物或其药学上可接受的盐,
[0011][0012]
其中,
[0013]
x代表-c(o)-或-s(o2)-;
[0014]
a1代表苯基、芳香杂环、取代苯基或取代芳香杂环;
[0015]
a2代表苯基、芳香杂环、取代苯基或取代芳香杂环;
[0016]
a1或a2中的取代基选自下述基团中的一种或几种:c
1-c6烷基、c
1-c6卤代烷基、c
1-c6烷氧基、苯基、取代苯基、羟基、硝基或卤素。
[0017]
在一种优选方案中,x代表-c(o)-。
[0018]
在一种优选方案中,a1代表下述基团:
[0019][0020]
r1或r2分别独立的代表氢原子、c
1-c4烷基、c
1-c4卤代烷基、c
1-c4烷氧基、苯基、羟基、硝基或卤素。
[0021]
在一种更优选方案中,r1代表氢原子、甲基、乙基、三氟甲基、三氯甲基、甲氧基、乙氧基、氟或氯。
[0022]
在一种特别优选方案中,r1代表氢原子或氯。
[0023]
在一种更优选方案中,r2代表氢原子、氟或氯。
[0024]
在一种特别优选方案中,r2代表氢原子。
[0025]
在一种优选方案中,a2代表下述基团:
[0026][0027]
r3或r4分别独立的代表氢原子、c
1-c4烷基、c
1-c4卤代烷基、c
1-c4烷氧基、苯基、羟基、硝基或卤素。
[0028]
在一种更优选方案中,r3代表氢原子、甲基、乙基、三氟甲基、三氯甲基、甲氧基、乙氧基、苯基、氟、氯、羟基或硝基。
[0029]
在一种特别优选方案中,r3代表氢原子、甲基、苯基、三氟甲基、氟或氯。
[0030]
在一种更优选方案中,r4代表氢原子、甲基、三氟甲基、苯基、氟或氯。
[0031]
在一种特别优选方案中,r4代表氢原子、甲基或苯基。
[0032]
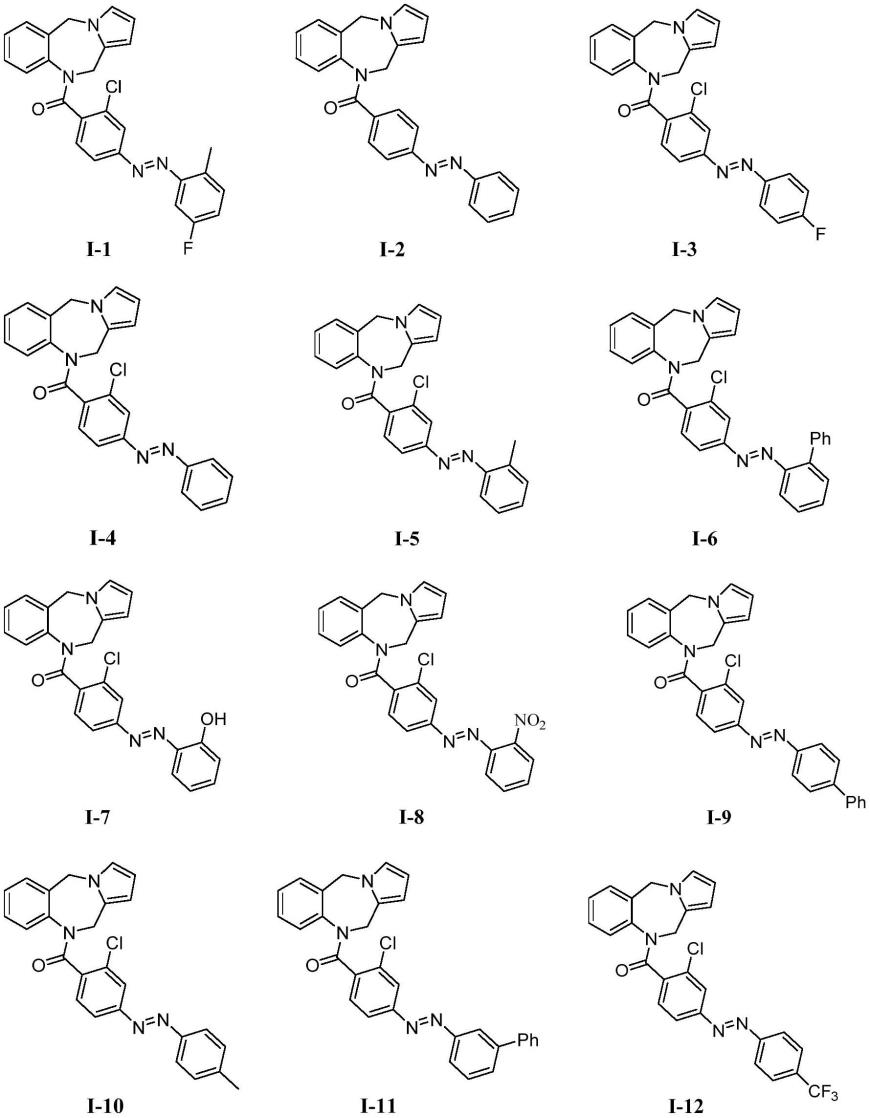
进一步地,在通式i所述化合物或其药学上可接受的盐中,所述化合物选自下列化合物:
[0033]
[0034][0035]
上述化合物的命名如下:
[0036]
(e)-(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)(2-氯-4-((5-氟-2-甲基苯基))二氮烯基)苯基)甲酮(化合物编号:i-1,下同);
[0037]
(e)-(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)(4-(苯基二氮烯基)苯基)甲酮(i-2);
[0038]
(e)-(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)(4-((4-氟苯基)二氮烯基)苯基)甲酮(i-3);
[0039]
(e)-(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)(2-氯-4-(苯基二氮烯基)苯基)甲酮(i-4);
[0040]
(e)-(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)(2-氯-4-(邻甲苯基二氮烯基)苯基)甲酮(i-5);
[0041]
(e)-(4-([1,1'-联苯]-2-基二氮烯基)-2-氯苯基)(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)甲酮(i-6;
[0042]
(e)-(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)(2-氯-4-((2-羟基苯基)二氮烯基)苯基)甲酮(i-7);
[0043]
(e)-(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)(2-氯-4-((2-硝基苯基)二氮烯基)苯基)甲酮(i-8);
[0044]
(e)-(4-([1,1'-联苯]-4-基二氮烯基)-2-氯苯基)(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)甲酮(i-9);
[0045]
(e)-(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)(2-氯-4-(对甲苯基二氮烯基)苯基)甲酮(i-10);
[0046]
(e)-(4-([1,1'-联苯基]-3-基二氮烯基)-2-氯苯基)(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)甲酮(i-11);
[0047]
(e)-(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)(2-氯-4-((4-(三氟甲基)苯基)二氮烯基))苯基)甲酮(i-12);
[0048]
(e)-(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)(2-氯-4-(间甲苯基二氮烯基)苯基)甲酮(i-13);
[0049]
(e)-(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)(2-氯-4-((4-氯苯基)二氮烯基)苯基)甲酮(i-14)。
[0050]
本发明还公开了通式i所示化合物的制备方法,x代表-c(o)-,a1或a2代表取代苯基时,它包括如下步骤:
[0051][0052]
其中,r1或r2为a1的取代基,r3或r4为a2的取代基。
[0053]
在一种优选方案中,通式i所示化合物的制备方法,它包括以下更加详细的步骤:邻硝基苄溴(化合物ii)与2-吡咯甲醛经取代反应得到中间体iii,再经氢气还原、环合得到
中间体iv。化合物v与不同取代的苯胺生成中间产物vi,中间产物vi经水解成对应的酸vii,再在二氯亚砜条件下制备对应的酰氯,再与中间产物iv生成酰胺,即得到通式i所示化合物。
[0054]
在由化合物ii制备中间体iii的反应中,所采用的溶剂选自n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dma)、乙腈、四氢呋喃、二氯甲烷、1,2-二氯乙烷或氯仿中的一种或多种,优先选自n,n-二甲基甲酰胺(dmf);采用的反应温度为0℃至室温。
[0055]
在由化合物iii制备iv或由化合物vi制备vii的反应中,所采用的溶剂选自乙醇、1,4-二氧六环、乙酸乙酯、二氯甲烷,优先选自乙醇;采用的反应温度为室温或回流。
[0056]
在由化合物v制备vi或者化合物vii的酰氯形式制备通式i化合物的反应中,所采用的溶剂选自二氯甲烷、1,4-二氧六环、乙酸乙酯、乙醇,优先选自二氯甲烷;采用的反应温度为0℃至室温。
[0057]
这些中间体或目标化合物均可按照常规分离技术加以纯化,并且根据需要将其转化为与可药用酸的加成盐。
[0058]
除非另外说明,在说明书和权利要求中使用的以下术语具有下面讨论的含义:
[0059]“药学上可接受的盐”表示保留母体化合物的生物有效性和性质的那些盐。这类盐包括:
[0060]
(1)与酸成盐,通过母体化合物的游离碱与无机酸或有机酸的反应而得,无机酸包括盐酸、氢溴酸、硝酸、磷酸、偏磷酸、硫酸、亚硫酸和高氯酸等,有机酸包括乙酸、三氟乙酸、丙酸、丙烯酸、己酸、环戊烷丙酸、羟乙酸、丙酮酸、草酸、(d)或(l)苹果酸、富马酸、马来酸、抗坏血酸、樟脑酸、苯甲酸、羟基苯甲酸、γ-羟基丁酸、甲氧基苯甲酸、邻苯二甲酸、甲磺酸、乙磺酸、萘-1-磺酸、萘-2-磺酸、对甲苯磺酸、水杨酸、酒石酸、柠檬酸、乳酸、肉桂酸、十二烷基硫酸、葡糖酸、谷氨酸、天冬氨酸、硬脂酸、扁桃酸、琥珀酸、戊二酸或丙二酸等。
[0061]
(2)存在于母体化合物中的酸性质子被金属离子代替或者与有机碱配位化合所生成的盐,金属例子例如碱金属离子、碱土金属离子或铝离子,有机碱例如乙醇胺、二乙醇胺、三乙醇胺、氨丁三醇、n-甲基葡糖胺、奎宁等。
[0062]“药物组合物”指将本发明中的化合物中的一个或多个或其药学上可接受的盐、溶剂化物、水合物或前药与别的化学成分,例如药学上可接受的载体,混合。药物组合物的目的是促进给药给动物的过程。
[0063]“药用载体”或“药学上可接受的载体”指的是对有机体不引起明显的刺激性和不干扰所给予化合物的生物活性和性质的药物组合物中的非活性成分,例如但不限于:碳酸钙、磷酸钙、各种糖(例如乳糖、甘露醇等)、淀粉、环糊精、硬脂酸镁、纤维素、碳酸镁、丙烯酸聚合物或甲基丙烯酸聚合物、凝胶、水、聚乙二醇、丙二醇、乙二醇、蓖麻油或氢化蓖麻油或多乙氧基氢化蓖麻油、芝麻油、玉米油、花生油等。
[0064]“烷基”表示1-20个碳原子的饱和的脂烃基,包括直链和支链基团(本技术书中提到的数字范围,例如“1-20”,是指该基团,此时为烷基,可以含1个碳原子、2个碳原子、3个碳原子等,直至包括20个碳原子)。更优选的是,烷基是有1-10个碳原子的中等大小的烷基,例如甲基、乙基、丙基、2-丙基、正丁基、异丁基、叔丁基、戊基等。最好是,烷基为有1-8或1-6个碳原子的低级烷基,例如甲基、乙基、丙基、2-丙基、正丁基、异丁基或叔丁基等。烷基可以是取代的或未取代的。当是取代的烷基时,该取代基优选是一或多个,更优选1-3个,最优选1
或2个取代基。
[0065]“芳香杂环”或“杂芳基”表示5至12个环原子的单环或稠合环基团,含有一个、两个、三个或四个选自n、o或s的环杂原子,其余环原子是c,另外具有完全共轭的π电子系统。未取代的杂芳基地非限制性实例有吡咯、呋喃、噻吩、咪唑、噁唑、噻唑、吡唑、吡啶、嘧啶、喹啉、异喹啉、嘌呤、四唑、三嗪和咔唑。杂芳基可以是取代的或未取代的。当被取代时,取代基优选为一个或多个,更为优选为一个、两个或三个,进而更为优选一个或两个,独立地选自以下基团,包括:低级烷基、三卤烷基、卤素、羟基、低级烷氧基、巯基、(低级烷基)硫基、氰基、酰基、硫代酰基、o-氨基甲酰基、n-氨基甲酰基、o-硫代氨基甲酰基、n-硫代氨基甲酰基、c-酰氨基、n-酰氨基、硝基、n-磺酰氨基、s-磺酰氨基。优选的杂芳基可选地被一个或两个取代基取代,取代基独立地选自卤素、低级烷基、三卤烷基、羟基、巯基、氰基、n-酰氨基、单或二烷基胺基、羧基或n-磺酰氨基。
[0066]
“‑
c(o)
‑”
表示co基团或基团。
[0067]
“‑
s(o2)
‑”
表示so基团或基团。
[0068]“羟基”表示-oh基团。
[0069]“硝基”表示-no2基团。
[0070]“烷氧基”表示-o-(未取代的烷基)和-o-(未取代的环烷基)。代表性实例包括但不限于甲氧基、乙氧基、丙氧基、丁氧基、环丙氧基、环丁氧基、环戊氧基、环己氧基等。
[0071]“卤素”表示氟、氯、溴或碘,优选为氟或氯。
[0072]“卤代烷基”表示卤素取代的烷基,优选如上所定义的卤素取代的低级烷基,它被一个或多个相同或不同的卤原子取代,例如-ch2cl、-cf3、-ch2cf3、-ch2ccl3等。
[0073]
本发明提供了一种药物组合物,它以本发明的化合物或其药学上可接受的盐为活性成分或主要活性成分,辅以药学上可接受的载体。
[0074]
本发明提供的上述苯并二氮卓并吡咯类化合物在制备精氨酸加压素受体拮抗剂药物方面的应用,特别是在制备与精氨酸加压素v2受体拮抗剂结合动力学药理工具相关药物方面的应用,更特别是在制备精氨酸加压素v2受体拮抗剂药物方面的应用。
[0075]
本发明的化合物或其药学上可接受的盐在制备与精氨酸加压素受体拮抗剂药物方面的应用,特别是在制备与精氨酸加压素v2受体拮抗剂结合动力学药理工具相关方面的应用,包括探索取代基大小与位置、特定光照波长等,调节配体构型,进而调控精氨酸加压素受体v2r的抑制状态,用于受体配体结合动力学的研究。
附图说明
[0076]
图1是365nm光照射(cis)或435nm光照射(trans)10min后i-10的药理学表征;
[0077]
图2是cis-i-10和trans-i-10的ki值随时间的变化,ki值与60min时的ki值有统计学差异,用***表示(p《0.001);
[0078]
图3是本发明部分化合物顺式和反式构型的动力学速率指数(kri),通过双点竞争结合实验获得数值,并由荧光标记配体结合度在60min时的值除以15min时的值得到;
[0079]
图4是cis-i-10和trans-i-10的动力学图谱,数据根据motulsky-mahan模型进行拟合;
[0080]
图5是cis-i-10和trans-i-10对camp生成的浓度依赖性的抑制作用;
[0081]
图6是空白组、cis-i-10和trans-i-10经多次洗脱后对精氨酸加压素诱导的camp产生的抑制作用。
具体实施方式
[0082]
通过以下实施例并结合附图对本发明的苯并二氮卓并吡咯类化合物作进一步的说明,但这些实施例不对本发明构成任何限制。
[0083]
实施例1
[0084]
(e)-(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)(2-氯-4-((5-氟-2-甲基苯基))二氮烯基)苯基)甲酮(i-1)
[0085][0086]
步骤1:将2-吡咯甲醛(475mg,5mmol)溶于dmf(15ml)中,冰浴条件下加入nah(180mg,7.5mmol),保持冰浴搅拌15min后加入邻硝基苄溴(1.08g,5mmol),室温搅拌过夜。向反应体系中加入大量水并充分搅拌,有棕色固体析出,减压抽滤,收集滤饼,得到浅棕色固体sn01005(1.1g,收率96%)。hrms(esi)c
12h11
n2o
3+
[m+h]
+
计算值:231.0764,实测值:231.0769.
[0087]
步骤2:将sn01005(500mg,2.2mmol),pd/c(150mg,10%钯,含55%水)置于茄形瓶中,加入乙醇(10ml),通入氢气,室温搅拌10h。减压抽滤除去钯碳,滤液减压浓缩,剩余物用flash柱层析分离纯化(乙酸乙酯/石油醚=1:20,v/v),得到白色固体sn01010(150mg,收率37%)。1h nmr(800mhz,chloroform-d)δ7.02(t,j=7.0hz,1h),6.99(d,j=7.5hz,1h),6.68(t,j=2.3hz,1h),6.62(t,j=7.4hz,1h),6.51(d,j=8.0hz,1h),6.04-6.01(m,2h),5.18(s,2h),4.47(s,2h).hrms(esi)c
12h13n2+
[m+h]
+
计算值:185.1073,实测值:185.1078.
[0088]
步骤3:将单过硫酸氢钾复合盐(oxone,6.15g,10mmol)溶于水(30ml),4-氨基-2-氯苯甲酸甲酯(925mg,5mmol)溶于二氯甲烷(20ml),再将后者滴入前者,并充分搅拌12h。有机层分别用无水硫代硫酸钠溶液,1n稀盐酸洗,再用无水硫酸钠干燥,浓缩;得到的油状物用30ml二氯甲烷溶解,再加入5-氟-2-甲基苯胺(676mg,5.4mmol),冰醋酸(6ml),室温搅拌过夜。二氯甲烷萃取,饱和食盐水洗涤,有机相用无水硫酸钠干燥,浓缩,剩余物用flash柱层析分离纯化(乙酸乙酯/石油醚=1:10,v/v),得到棕黄色固体sn01078(860mg,收率56%)。1h nmr(800mhz,dmso-d6)δ8.03(d,j=8.2hz,1h),7.99(d,j=1.8hz,1h),7.96-7.93(m,1h),7.54-7.50(m,1h),7.41-7.37(m,1h),7.37-7.34(m,1h),3.91(s,3h),2.67(s,3h).hrms(esi)c
15h13
clfn2o
2+
[m+h]
+
计算值:307.0644,实测值:307.0654.
[0089]
步骤4:将sn01078(1.13g,3.7mmol)与乙醇(15ml)混合,再加入氢氧化钾(400mg,
7.1mmol),水(2ml),回流1h。待反应液冷却至室温,用稀盐酸(4n)调ph至4-6,有大量固体析出,减压抽滤,收集滤饼,得到棕红色固体sn01078a。不经纯化,直接用于下一步。
[0090]
步骤5:将sn01078a(258mg,0.88mmol)溶于二氯亚砜(3ml),再滴入1滴dmf,回流1h,减压除去溶剂备用。将sn01010(100mg,0.54mmol)溶于无水二氯甲烷(5ml),再加入三乙胺(0.24ml,1.8mmol),冰浴条件下将制得的酰氯用无水二氯甲烷转移至反应液中,保持冰浴30min后室温搅拌30min。二氯甲烷萃取,饱和食盐水洗涤,有机相用无水硫酸钠干燥,浓缩,剩余物用flash柱层析分离纯化(乙酸乙酯/石油醚=1:10,v/v),得到黄色固体i-1(89mg,收率36%)。1h nmr(800mhz,dmso-d6)δ7.84(s,1h),7.72(s,1h),7.62(s,1h),7.52-7.47(m,1h),7.41(d,j=7.1hz,1h),7.38-7.34(m,1h),7.33-7.28(m,1h),7.16-7.05(m,3h),6.87-6.82(m,1h),6.02(s,1h),5.93-5.90(m,1h),5.49-5.07(m,3h),2.63(s,3h).
13
c nmr(201mhz,dmso-d6)δ165.96,160.82(d,j=244.4hz),152.05,150.24(d,j=5.3hz),138.46,135.57,134.87,134.86,133.12,133.08,128.78,128.67,127.95,124.86,122.70,122.22,121.69,118.95,118.84,108.60,106.84,101.63,101.52,49.40,45.09,16.36.hrms(esi)c
26h21
fcln4o
+
[m+h]
+
计算值:459.1382,实测值:459.1376.hplc:98.8%。
[0091]
实施例2
[0092]
(e)-(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)(4-(苯基二氮烯基)苯基)甲酮(i-2)
[0093][0094]
步骤1-3:仿照实施例1所述步骤3-5的方法,将其步骤3中“4-氨基-2-氯苯甲酸甲酯”替换为“对氨基苯甲酸甲酯”,“5-氟-2-甲基苯胺”替换为“苯胺”,其余所需原料、试剂相同,得到橙色固体i-2(40mg,收率19%)。1h nmr(800mhz,dmso-d6)δ7.89-7.82(m,2h),7.71(d,j=7.2hz,2h),7.61-7.56(m,3h),7.52-7.45(m,3h),7.17(t,j=7.0hz,1h),7.08(s,1h),6.96(s,1h),6.87-6.83(m,1h),6.02-5.96(m,1h),5.93(t,j=2.8hz,1h),5.33(s,3h).
13
c nmr(201mhz,dmso-d6)δ168.66,151.93,151.85,141.63,138.40,135.13,131.89,129.47(2c),129.30,129.15(2c),128.90,128.77,127.56,125.37,122.64(2c),122.08(2c),121.91,108.04,106.83,49.65,45.61.hrms(esi)c
25h21
n4o
+
[m+h]
+
计算值:393.1710,实测值:393.1708.hplc:98.5%。
[0095]
实施例3
[0096]
(e)-(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)(4-((4-氟苯基)二氮烯基)苯基)甲酮(i-3)
[0097][0098]
步骤1-3:仿照实施例1所述步骤3-5的方法,将其步骤3中“5-氟-2-甲基苯胺”替换为“4-氟苯胺”,其余所需原料、试剂相同,得到橙色固体i-3(70mg,收率29%)。1h nmr(800mhz,dmso-d6)δ7.97-7.94(m,2h),7.79(s,1h),7.68(s,1h),7.65-7.58(m,1h),7.45(t,j=8.7hz,2h),7.41(d,j=7.3hz,1h),7.15-7.05(m,3h),6.87-6.83(m,1h),6.07-6.00(m,1h),5.92(t,j=3.0hz,1h),5.59-4.94(m,4h).
13
c nmr(201mhz,dmso-d6)δ166.01,164.90,163.65,151.96,148.42,148.41,138.18,135.58,128.74,128.61,127.92,125.33,125.28,124.86(2c),122.22(2c),122.08,121.69,116.62,116.51,108.63,106.84,49.39,45.08.hrms(esi)c
25h19
fcln4o
+
[m+h]
+
计算值:445.1226,实测值:445.1220.hplc:98.2%。
[0099]
实施例4
[0100]
(e)-(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)(2-氯-4-(苯基二氮烯基)苯基)甲酮(i-4)
[0101][0102]
步骤1-3:仿照实施例1所述步骤3-5的方法,将其步骤3中“5-氟-2-甲基苯胺”替换为“苯胺”,其余所需原料、试剂相同,得到橙色固体i-4(40mg,收率17%)。1h nmr(800mhz,dmso-d6)δ7.90-7.85(m,2h),7.80(s,1h),7.69(s,1h),7.66-7.62(m,1h),7.61-7.59(m,3h),7.41(d,j=7.2hz,1h),7.12(t,j=7.3hz,1h),7.11-7.05(m,2h),6.86-6.83(m,1h),6.05-6.01(m,1h),5.92(t,j=3.0hz,1h),5.61-4.88(m,4h).
13
c nmr(201mhz,dmso-d6)δ166.01,152.07,151.61,138.17,135.57,132.31,129.59,129.51(2c),128.74,128.63,127.91,124.86,122.90,122.83(2c),122.65,122.45,122.21,122.10,121.68,108.62,106.84,49.40,45.08.hrms(esi)c
25h20
cln4o
+
[m+h]
+
计算值:427.1320,实测值:427.1330.hplc:97.7%。
[0103]
实施例5
[0104][0105]
(e)-(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)(2-氯-4-(邻甲苯基
7.54(m,1h),7.45-7.42(m,1h),7.40(d,j=7.1hz,1h),7.14-7.08(m,2h),7.08-7.04(m,2h),6.95(t,j=8.1hz,1h),6.87
–
6.83(m,1h),6.05
–
5.99(m,1h),5.92(t,j=3.0hz,1h),5.31(s,4h).
13
c nmr(201mhz,dmso-d6)δ166.14,155.71,151.98,138.58,137.74,135.57,134.61,128.73,128.65,127.92,124.90,122.51,122.47,122.25,122.21,121.86,120.37,120.34,119.82,119.75,118.47,108.61,106.84,49.41,45.07.hrms(esi)c
25h20
cln4o
2+
[m+h]
+
计算值:443.1269,实测值:443.1271.hplc:》99.0%。
[0115]
实施例8
[0116][0117]
(e)-(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)(2-氯-4-((2-硝基苯基)二氮烯基)苯基)甲酮(i-8)
[0118]
步骤1-3:仿照实施例1所述步骤3-5的方法,将其步骤3中“5-氟-2-甲基苯胺”替换为“2-硝基苯胺”,其余所需原料、试剂相同,得到橙色固体i-8(97mg,收率38%)。1h nmr(800mhz,dmso-d6)δ8.14(d,j=7.2hz,1h),7.85(t,j=7.7hz,1h),7.81-7.75(m,2h),7.72-7.61(m,3h),7.41(d,j=7.3hz,1h),7.16-7.06(m,3h),6.88-6.83(m,1h),6.04-6.02(m,1h),5.92(t,j=3.0hz,1h),5.32(s,4h).
13
c nmr(201mhz,dmso-d6)δ165.79,157.28,151.82,147.04,143.87,139.27,137.97,135.60,133.88,132.41,128.81,128.72,128.03,124.80,124.36,123.00,122.27,122.25,121.76,118.51,118.40,108.64,106.85,49.39,45.10.hrms(esi)c
25h19
cln5o
3+
[m+h]
+
计算值:472.1180,实测值:472.1171.hplc:97.7%。
[0119]
实施例9
[0120]
(e)-(4-([1,1'-联苯]-4-基二氮烯基)-2-氯苯基)(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)甲酮(i-9)
[0121][0122]
步骤1-3:仿照实施例1所述步骤3-5的方法,将其步骤3中“5-氟-2-甲基苯胺”替换为“4-氨基联苯”,其余所需原料、试剂相同,得到淡黄色固体i-9(140mg,收率52%)。1h nmr(800mhz,dmso-d6)δ7.96(d,j=8.5hz,2h),7.91(d,j=8.5hz,2h),7.86-7.79(m,1h),7.78(d,j=7.4hz,2h),7.70(s,1h),7.65-7.56(m,1h),7.51(t,j=7.7hz,2h),7.45-7.41(m,2h),7.16-7.10(m,2h),7.08(t,j=7.5hz,1h),6.89-6.84(m,1h),6.06-6.03(m,1h),5.94(t,j=3.0hz,1h),5.58-4.89(m,4h).
13
c nmr(201mhz,dmso-d6)δ166.00,152.17,150.76,143.71,139.67,138.72,138.10,135.54,130.54,130.49,129.05(2c),128.72,128.32,
127.91,127.74,127.66(2c),126.87(2c),124.87,123.60,123.52(2c),122.19,122.10,121.66,108.59,106.82,49.40,45.06.hrms(esi)c
31h24
cln4o
+
[m+h]
+
计算值:503.1626,实测值:503.1633.hplc:98.9%。
[0123]
实施例10
[0124]
(e)-(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)(2-氯-4-(对甲苯基二氮烯基)苯基)甲酮(i-10)
[0125][0126]
步骤1-3:仿照实施例1所述步骤3-5的方法,将其步骤3中“5-氟-2-甲基苯胺”替换为“4-甲基苯胺”,其余所需原料、试剂相同,得到橙色固体i-10(83mg,收率35%)。1h nmr(800mhz,dmso-d6)δ7.77(d,j=8.0hz,3h),7.65(s,1h),7.62-7.53(m,1h),7.40(t,j=8.9hz,3h),7.14-7.01(m,3h),6.88-6.82(m,1h),6.05-6.00(m,1h),5.96-5.89(m,1h),5.56-4.92(m,4h),2.39(s,3h).
13
c nmr(201mhz,dmso-d6)δ166.03,152.14,149.76,142.78,137.87,135.52,133.04,130.07,130.00(2c),128.70,128.60,127.86,126.17,124.87,122.86(2c),122.21,122.18,121.93,121.52,108.58,106.81,49.39,45.05,21.05.hrms(esi)c
26h22
cln4o
+
[m+h]
+
计算值:441.1477,实测值:441.1482.hplc:96.1%。
[0127]
实施例11
[0128]
(e)-(4-([1,1'-联苯基]-3-基二氮烯基)-2-氯苯基)(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)甲酮(i-11)
[0129][0130]
步骤1-3:仿照实施例1所述步骤3-5的方法,将其步骤3中“5-氟-2-甲基苯胺”替换为“3-氨基联苯”,其余所需原料、试剂相同,得到橙色泡沫状固体i-11(105mg,收率39%)。1h nmr(800mhz,dmso-d6)δ8.13(s,1h),7.89(d,j=7.6hz,1h),7.86-7.81(m,2h),7.75(d,j=7.5hz,2h),7.73-7.70(m,1h),7.67(t,j=7.8hz,1h),7.60-7.55(m,1h),7.50(t,j=7.6hz,2h),7.44-7.39(m,2h),7.14-7.04(m,3h),6.85(s,1h),6.05-6.01(m,1h),5.93(t,j=2.9hz,1h),5.31(s,4h).
13
c nmr(201mhz,dmso-d6)δ165.99,152.19,152.07,141.46,138.97,138.25,138.24,135.61,135.56,133.04,130.36,130.11,130.09,129.04(2c),128.72,128.66,128.01,127.89,126.82,126.80(2c),126.20,124.86,122.20,122.17,121.74,121.46,121.15,49.40,45.06.hrms(esi)c
31h24
cln4o
+
[m+h]
+
计算值:503.1633,实测值:503.1637.hplc:98.9%。
[0131]
实施例12
[0132]
(e)-(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)(2-氯-4-((4-(三氟
甲基)苯基)二氮烯基))苯基)甲酮(i-12)
[0133][0134]
步骤1-3:仿照实施例1所述步骤3-5的方法,将其步骤3中“5-氟-2-甲基苯胺”替换为“4-三氟甲基苯胺”,其余所需原料、试剂相同,得到橙黄色固体i-12(128mg,收率48%)。1h nmr(800mhz,dmso-d6)δ8.02(d,j=8.3hz,2h),7.96(d,j=8.4hz,2h),7.88-7.83(m,1h),7.77-7.71(m,1h),7.69-7.60(m,1h),7.41(d,j=7.2hz,1h),7.14-7.04(m,3h),6.87-6.83(m,1h),6.05-6.02(m,1h),5.92(t,j=3.0hz,1h),5.32(s,4h).
13
c nmr(201mhz,dmso-d6)δ165.87,153.65,151.85,138.89,135.58,131.45,128.74,128.62,127.95,126.72,126.70,124.82,124.47,123.36(2c),123.12,122.45,122.21(2c),122.02,116.32,112.82,108.62,106.82,49.38,45.07.hrms(esi)c
26h19
f3cln4o
+
[m+h]
+
计算值:495.1194,实测值:495.1187.hplc:97.7%。
[0135]
实施例13
[0136]
(e)-(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)(2-氯-4-(间甲苯基二氮烯基)苯基)甲酮(i-13)
[0137][0138]
步骤1-3:仿照实施例1所述步骤3-5的方法,将其步骤3中“5-氟-2-甲基苯胺”替换为“3-甲基苯胺”,其余所需原料、试剂相同,得到橙黄色固体i-13(40mg,收率17%)。1h nmr(800mhz,dmso-d6)δ7.78(s,1h),7.70-7.66(m,3h),7.59-7.54(m,1h),7.49(t,j=7.9hz,1h),7.41(t,j=8.0hz,2h),7.15-7.04(m,3h),6.88-6.82(m,1h),6.04-6.01(m,1h),5.92(t,j=3.0hz,1h),5.48-5.09(m,3h),2.41(s,3h).
13
c nmr(201mhz,dmso-d6)δ166.03,152.11,151.71,139.06,138.09,135.58,134.45,133.01,132.95,129.37,129.31,128.74,128.62,127.91,124.86,122.80,122.21,122.10,122.02,121.65,120.53,108.62,106.84,49.39,45.08,20.81.hrms(esi)c
26h22
cln4o
+
[m+h]
+
计算值:441.1477,实测值:441.1486.hplc:98.4%。
[0139]
实施例14
[0140]
(e)-(5h-苯并[e]吡咯并[1,2-a][1,4]二氮杂-10(11h)-基)(2-氯-4-((4-氯苯基)二氮烯基)苯基)甲酮(i-14)
[0141][0142]
步骤1-3:仿照实施例1所述步骤3-5的方法,将其步骤3中“5-氟-2-甲基苯胺”替换为“4-氯苯胺”,其余所需原料、试剂相同,得到橙黄色固体i-14(84mg,收率34%)。1h nmr(800mhz,dmso-d6)δ7.99-7.96(m,1h),7.89(d,j=8.7hz,2h),7.81(s,1h),7.73-7.68(m,1h),7.67(d,j=8.7hz,2h),7.40(d,j=7.3hz,1h),7.12(t,j=7.4hz,1h),7.10-7.05(m,2h),6.86-6.83(m,1h),6.02(s,1h),5.92(t,j=3.0hz,1h),5.32(s,4h).
13
c nmr(201mhz,dmso-d6)δ165.97,151.91,150.16,138.41,136.87,135.58,129.75,129.67(2c),129.21,128.75,128.62,127.93,124.84,124.65,124.57,124.50(2c),122.22,122.21,121.81,108.63,106.84,49.39,45.08.hrms(esi)c
25h19
cl2n4o
+
[m+h]
+
计算值:461.0930,实测值:461.0941.hplc:98.2%。
[0143]
下面是本发明的代表化合物的部分药理试验及结果:
[0144]
通过光照,光照条件为365nm。配体分子的构型发生顺式到反式异构化(例如,反式构型为本发明中产物i-10的结构,异构化过程如下所示)。配体分子的活性也随之发生变化,产生可逆的光照前后不同构型的活性差异。
[0145][0146]
1、受体亲和力性质表征
[0147]
分别对实施例1-13中的光控配体进行亲和力实验。具体方案如下:
[0148]
将目标化合物(1
×
10-4
m-1
×
10-10
m)、conivaptan(100μm)、tag-lite缓冲液(1
×
)(1%dmso)、v2r荧光配体(6.3nm)依次加入384孔板中。然后与snap标记的hek293-hv2r细胞在37℃孵育1h,多功能酶标仪在620nm和665nm处记录荧光强度,软件分析后获得亲和力值。测试结果见表1:
[0149]
表1光控配体对受体v2的亲和力
[0150][0151]
备注:pas即光诱导亲和力漂移(photo-induced affinity shift),由反式构型(trans)的ki值除以顺式构型(cis)的ki值得到。n.d.为不适用。
[0152]
结果表明,在所测定的化合物中,顺式的i-2、i-4、i-10与i-13的受体亲和力较强,其ki值均小于200nm,顺式与反式构型间的亲和力漂移分别为6.6倍、3.1倍、16.5倍与2.7倍。其中i-10经紫外光照后的顺式构型为优势构型,对v2受体亲和力最强(ki为17nm),而其热力学稳定的反式构型的ki在数值上是顺式构型的16.5倍(其结合曲线见附图1),是所测定化合物中亲和力漂移最大的,实现了紫外光照前后活性的off-on切换。此外,结合不同孵育时间ki的变化情况(附图2),顺式i-10孵育10min与60min的ki值有显著差异而反式无明显差异,表明其顺反式构型在结合动力学上可能具有不同。
[0153]
2、动力学性质表征
[0154]
分别对实施例1,3-6、9-13中的光控配体进行动力学性质测定。具体方案如下:将目标化合物(ic
50
)、conivaptan(100μm)、tag-lite缓冲液(1
×
)(1%dmso)、v2r荧光配体(6.3nm)依次加入384孔板中。在37℃下,立即用多功能酶标仪在620nm和665nm处连续测量1h(或测量15和60min的值),记录荧光强度,软件分析后获得kri、k
on
、k
off
和rt值。
[0155]
测试结果如表2-3及附图3-4所示:
[0156]
表2动力学性质初筛结果
[0157][0158]
备注:kri即动力学速率指数(kinetic rate index)的缩写,由双点竞争性结合实验获得,kri《1代表停留时间较短,kri》1代表停留时间较长。
[0159]
结合表2与图3可以看出,i-3、i-6、i-9热力学稳定的反式构型与i-4、i-5、i-9、i-10紫外光照后的顺式构型的kri大于1(其中i-10的kri值最大,为1.72),表明这些构型的化合物的停留时间较长;而这些化合物的顺式与反式构型kri的差值中,仅i-6、i-9和i-10的差值大于等于0.10(其中i-10的差值最大,为0.77),表明两种构型之间的停留时间有不同。其中i-10的顺式构型的kri在系列化合物中最大,数值上远大于1,而其反式构型的kri小于1,此数据提示,i-10的顺反构型之间存在较大的停留时间上的差异,具有进一步研究的价值。
[0160]
表3化合物i-10(sn02010)的动力学指标
[0161][0162]
结合表3与图4可以看出,化合物i-10(sn02010)的顺式与反式构型在结合动力学上具有较大差异,其顺式构型的停留时间是反式构型的4.3倍,趋势上与ki值的差异相近,具有作为药理学研究工具的潜在价值。
[0163]
3、camp实验
[0164]
对实施例i-10(sn02010)中光控配体进行camp实验。具体方案如下:
[0165]
功能活性试验:在含有hepes(5mm)、0.1%(w/v)bsa、cilostamide(50μm)、
rolipram(50μm)和腺苷脱氨酶(ada)(0.8iu/ml)的实验缓冲液中,血管加压素v2r的配体可调节camp的积累。
[0166]
chohv2r细胞(1000个细胞/孔)加入i-10(cis/trans)(1
×
10-4
m-1
×
10-10
m)在37℃下反应1h。加入抗利尿激素(avp)的混合物,在37℃条件下作用1h。细胞camp水平由cisbio生物公司(上海)的camp-gi试剂盒测定。
[0167]
洗脱实验:chohv2r细胞加入i-10(cis/trans)(100
×
ki),在37℃条件下作用1h。离心,弃去原有溶液,用空白的实验缓冲液重悬,在37℃条件下孵育10min。再次离心,并弃去原有溶液,空白缓冲液重悬,在37℃条件下孵育另一个10min。重悬为1000个细胞/孔,加入抗利尿激素(avp)的混合物,在37℃条件下作用1h。细胞camp水平由cisbio生物公司(上海)的camp-gi试剂盒测定。
[0168]
测试结果如图5-6所示:
[0169]
功能活性实验结果表明(图5),代表化合物i-10的顺式构型的功能活性明显强于反式构型,这与它们的结合亲和力相一致;洗脱实验结果表明(图6),将未结合配体经完全的洗脱处理后,顺式i-10的camp水平仅恢复了30%,而反式i-10的camp水平则完全恢复。
[0170]
以上药理学数据显示,本发明化合物(特别是i-10)可以在光的调控下,以高度的时空分辨率对v2r的活性进行调控,从而可作为新的v2r药理学研究工具加以应用。
[0171]
以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可能对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1