一种阿维巴坦钠中间体的连续流合成方法与流程
1.本发明涉及药物化学合成技术领域,具体涉及一种阿维巴坦钠中间体的连续流合成方法。
背景技术:
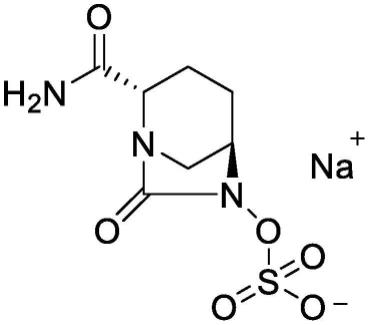
2.阿维巴坦钠,中文名:(2s,5r)-6-(磺基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺钠盐,结构式如下所示:
[0003][0004]
阿维巴坦,是novexel公司研发的一种新型的β-内酰胺酶抑制剂),于2011年获得专利,2015年2月actavis首次在fda上市,国内商品名思福妥。国内市场已上市三种类似机理药物,与同为β-内酰胺酶抑制剂(舒巴坦、他唑巴坦、克拉维酸)相比,作用时间长;与β-内酰胺酶形成可逆性共价结合;且不会诱导β-内酰胺酶产生等优点。与其他β-内酰胺类抗生素(例如:头孢和碳青霉烯抗生素)联合使用时,具有广谱抗菌活性,尤其是对β-内酰胺类抗生素耐药性强的大肠杆菌和克雷伯肺炎杆菌,具有抗菌活性显著。2012年,dubreui等研究发现,针对316起厌氧菌菌株的抑菌性,头孢他啶-阿维巴坦-甲硝唑三药联用(8:8:1)抑制了290起,实验显现出了高效的抑菌活性,三药联用法对厌氧菌菌株抑菌性高达91.8%。对大多数菌株均显示出增效或协同作用,且未发现菌株耐药。因此,阿维巴坦钠的新合成路线的研究具有很大的应用价值。
[0005][0006]
cn1468242和cn102834395公开了,合成阿维巴坦的方法,其合成路线如下:化合物专利cn1289500c及cn103649051b、cn105294690b公开了
[0007][0008]
化合物专利cn107417686a公开了一种阿维巴坦纳的合成方法,其合成路线如下:
[0009][0010]
在研究合成化合物ii路线中,参考众多专利,经整理化合物ii作为阿维巴坦钠合成的关键中间体,其方法均需要经使用催化加氢的方式,来合成重要中间体阿维巴坦钠化合物ii,合成方法均使用传统间歇式压力氢化反应釜进行氢化合成。生产中传统的间歇式压力氢化反应釜难以避免的会出现,耗时较长,转化率较低,杂质多,且存在安全性风险。
技术实现要素:
[0011]
本发明的目的在于提供一种适用于未来工业实现连续化、自动化的新氢化合成方法,优化传统氢化工艺,发明了一种阿维巴坦钠中间体的连续流氢化合成方法,采用连续流技术使用连续微反应加氢反应器,催化氢化阿维巴坦钠中间体i制备阿维巴坦钠中间体ii。该方法与常规的间歇式压力氢化反应釜催化氢化工艺相比,避免了加氢过程高压氢气和反应时间长等问题,从而更加确保生产过程中的安全性;该工艺反应时间短,持液体积小,无放大效应,本质上改变了反应的安全性;相比间歇式反应避免了反应出料的危险过程,更易于实现工业生产连续化、自动化;目标产物转化率在90%以上,纯度在99%以上,有利于现代工业化。
[0012]
本发明的技术解决方案如下:
[0013]
一种阿维巴坦钠中间体的连续流合成方法,将阿维巴坦钠中间体i与催化剂和质子性溶剂及缚酸剂的混合溶液作为物料一,将氢气作为物料二,通过连续微反应加氢反应器中进行氢化反应,在合适的条件连续流氢化合成阿维巴坦钠中间体ii;其合成路线如下所示:
[0014]
[0015]
其中,r1代表:苄基;
[0016]
其中,r2季铵盐:代表四辛基溴化铵、四己基溴化铵、三辛基甲基溴化铵、三丁基己基溴化铵;
[0017]
其中,所述阿维巴坦钠中间体i为(2s,5r)-6-(苄基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺,其中,r1代表:苄基;
[0018]
其中,所述阿维巴坦钠中间体ii为(2s,5r)-6-(磺基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺正四丁基季铵盐正离子、四辛基铵季铵盐正离子、三辛基甲基铵季铵盐正离子;其中,r2季铵盐:正四丁基季铵盐正离子、四辛基铵季铵盐正离子、三辛基甲基铵季铵盐正离子;
[0019]
进一步的,反应中,所述的催化剂为钯炭或氢氧化钯中的一种或其中两种配合使用,优选钯炭与氢氧化钯配合使用;优选钯炭与氢氧化钯质量比为1:1。
[0020]
进一步的,反应中,所述钯炭催化剂的钯负载量为5-10wt%,优选钯负载量为10wt%,
[0021]
进一步的,反应中,所述阿维巴坦钠中间体i与催化剂量的质量比,比例不限于1:0.06-0.15,优选比例为1:0.06-0.1。
[0022]
进一步的,反应中,所述的缚酸剂包括但不限于三乙胺、n,n-二异丙基乙胺、吡啶。优选为n,n-二异丙基乙胺。
[0023]
进一步的,反应中,所述质子类溶剂包括但不限于甲醇、乙醇、异丙醇、水,溶剂可能为其中一种或其中两种,优选为异丙醇、水;其两种溶剂比例,比例不限于1:1-10,其水和异丙醇和优选比例为1:7。
[0024]
进一步的,反应中,所述阿维巴坦钠中间体i与单一或混合溶剂的质量比,比例不限于1:1-20,优选中间体i与溶剂质量比为1:20,更优选为1:12。
[0025]
进一步的,所述微通道氢化反应器包括流路一、流路二、流路三、预热模块、反应模块以及气液分离装置,所述的物料一经流路一依序经过预热模块、反应模块和气液分离装置,所述的物料二经流路二进入反应模块,流路三中分离出的溶剂、催化剂和缚酸剂,返回流路一中重新利用。
[0026]
进一步的,反应中,所述物料一的流速为50-150g/min,优选为80g/min;物料二的流速的为标况(1个标准大气压状况)下600-2000ml/min,优选为1100ml/min。
[0027]
进一步的,反应中,所述预热模块的温度在40-80℃,所述的反应温度是指反应模块中温度在40-80℃,优选为60℃;所述的反应压力为15-100bar,优选为20bar。
[0028]
进一步的,反应中,所述反应时间为60-350s,优选为100s。
[0029]
与现有间歇性氢化技术相比,本发明具备如下有益效果:
[0030]
1)与传统的间歇式压力氢化反应釜相比,由于采用连续微反应加氢反应器进行氢化合成,反应过程持液体积小,降低了氢化反应的安全隐患,大大提升了反应、生产的安全性;该工艺反应时间短,反应持液体积小,无放大效应,本质上改变了反应的安全性;反应液中产物纯度在90%以上;反应液经催化剂分离、有机溶剂萃取、调ph值、配离子对成盐、有机溶剂萃取、洗涤、浓缩、脱色、套蒸、结晶、干燥等后处理步骤得到的相关产品(阿维巴坦钠中间体ii)摩尔收率在90%以上,产品纯度在99.0%以上,有利于工业化生产。
[0031]
2)本发明使用了缚酸剂,减少h
+
对钢铁的腐蚀,更适用于钢铁材质的微通道反应
器。
[0032]
3)本发明反应时间大大缩短,从间歇式压力氢化反应釜反应的5-24小时,缩短至60至350s。
[0033]
4)本发明的工艺通过多组反应模块试验结果显示无放大效应,为阿维巴坦钠中间体i制备阿维巴坦钠中间体ii提供了连续流反应新方法。
[0034]
5)本发明利用了微反应器混合高效和优异的传质传热性能,强化了反应过程中的相间传质和移热能力,可以显著减小反应器体积,提高反应收率,提高生产效率和安全性。该方法可以解决间歇式压力氢化反应釜工艺中的生产效率低,反应纯度差以及装置危险性大等问题,可以实现过程的连续自动化操作,具有收率高和安全性好等优点。
附图说明
[0035]
图1为阿维巴坦钠中间体i的连续流合成工艺流程。
具体实施方式
[0036]
以下结合附图,更清楚地理解本发明的目的、特征和优点,及具体实施例对本发明作进一步阐释。如无特别说明,所述原材料均能从商业途径获得;采用高通量微通道氢化反应器g1-1型;美国康宁公司。
[0037]
实施例1:(2s,5r)-6-(磺基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺正四丁基季铵盐的制备
[0038]
1、装置:连续流微通道氢化反应器,参照图1确定微通道连接模式,混合反应模块,根据流速与反应停留时间确定,换热介质为乙醇;
[0039]
2、如图1所示,流路一:料液配制,50.0g化合物(i)、3.0g氢氧化钯催化剂、180ml水、420ml异丙醇,搅拌,形成混悬液,通过料泵打入微通道反应器,通过计量模块设置流速为50.0g/min。化合物(i)为:(2s,5r)-6-(苄基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺。
[0040]
3、流路二:物料氢气,通过气路控制器,设置气体流速为1100ml/min;
[0041]
4、管路压力控制:氮气备压管路压力至20bar;
[0042]
5、将系统设置循环温度60℃,通过连续流反应器8块反应模块(204ml体积)反应300秒后,收集反应液。反应液中(2s,5r)-6-(羟基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺的纯度为95.33%。过滤脱除反应液中钯炭,转入磺化釜进行磺酸化(磺酸化试机为三氧化硫的络合物),经乙酸丁酯有机相萃取,离子对成盐(铵离子源为正四丁基季铵盐正离子),二氯甲烷萃取产物,无水硫酸钠干燥后,经减压浓缩,用4-甲基-2-戊酮进行套蒸析晶,过滤,使用4-甲基-2-戊酮淋洗,减压烘干得(2s,5r)-6-(磺基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺正四丁基季铵盐。液相色谱纯度99.99%,最大单杂未检出,摩尔收率91.11%。
[0043]
实施例2:(2s,5r)-6-(磺基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺正四辛基铵季铵盐的制备
[0044]
1、装置:连续流微通道氢化反应器,参照图1确定微通道连接模式,混合反应模块,根据流速与反应停留时间确定,换热介质为乙醇;
[0045]
2、如图1所示,流路一:料液配制,50.0g化合物(i)、3.0g氢氧化钯催化剂、180ml水、420ml异丙醇,搅拌,形成混悬液,通过料泵打入微通道反应器,通过计量模块设置流速为50.0g/min。化合物(i)为:(2s,5r)-6-(苄基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺。
[0046]
3、流路二:物料氢气,通过气路控制器,设置气体流速为1100ml/min;4、管路压力控制:氮气备压管路压力至20bar;
[0047]
5、将系统设置循环温度60℃,通过连续流反应器8块反应模块(204ml体积)反应300秒后,收集反应液。反应液中(2s,5r)-6-(羟基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺的纯度为95.13%。过滤脱除反应液中钯炭,转入磺化釜进行磺酸化(磺酸化试机为三氧化硫的络合物),经乙酸丁酯有机相萃取,离子对成盐(铵离子源为四辛基铵季铵盐正离子),二氯甲烷萃取产物,无水硫酸钠干燥后,经减压浓缩,用4-甲基-2-戊酮进行套蒸析晶,过滤,使用4-甲基-2-戊酮淋洗,减压烘干得(2s,5r)-6-(磺基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺正四辛基铵季铵盐。液相色谱纯度99.99%,最大单杂未检出,摩尔收率92.15%。
[0048]
实施例3:(2s,5r)-6-(磺基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺正三辛基甲基铵季铵盐的制备
[0049]
1、装置:连续流微通道氢化反应器,参照图1确定微通道连接模式,混合反应模块,根据流速与反应停留时间确定,换热介质为乙醇;
[0050]
2、如图1所示,流路一:料液配制,50.0g化合物(i)、3.0g氢氧化钯催化剂、180ml水、420ml异丙醇,搅拌,形成混悬液,通过料泵打入微通道反应器,通过计量模块设置流速为50.0g/min。化合物(i)为:(2s,5r)-6-(苄基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺。
[0051]
3、流路二:物料氢气,通过气路控制器,设置气体流速为1100ml/min;4、管路压力控制:氮气备压管路压力至20bar;
[0052]
5、将系统设置循环温度60℃,通过连续流反应器8块反应模块(204ml体积)反应300秒后,收集反应液。反应液中(2s,5r)-6-(羟基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺的纯度为95.82%。过滤脱除反应液中钯炭,转入磺化釜进行磺酸化(磺酸化试机为三氧化硫的络合物),经乙酸丁酯有机相萃取,离子对成盐(铵离子源为正三辛基甲基铵季铵盐正离子),二氯甲烷萃取产物,无水硫酸钠干燥后,经减压浓缩,用4-甲基-2-戊酮进行套蒸析晶,过滤,使用4-甲基-2-戊酮淋洗,减压烘干得(2s,5r)-6-(磺基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺正三辛基甲基铵季铵盐。液相色谱纯度99.99%,最大单杂未检出,摩尔收率93.25%。
[0053]
实施例4:(2s,5r)-6-(磺基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺正三辛基甲基铵季铵盐的制备
[0054]
1、装置:连续流微通道氢化反应器,参照图1确定微通道连接模式,混合反应模块,根据流速与反应停留时间确定,换热介质为乙醇;
[0055]
2、如图1所示,流路一:料液配制,50.0g化合物(i)、1.5g10wt%钯炭、1.5g氢氧化钯催化剂、180ml水、420ml异丙醇,搅拌,形成混悬液,通过料泵打入微通道反应器,通过计量模块设置流速为50.0g/min。化合物(i)为:(2s,5r)-6-(苄基氧基)-7-氧代-1,6-二氮杂
二环[3.2.1]辛烷-2-甲酰胺。
[0056]
3、流路二:物料氢气,通过气路控制器,设置气体流速为1100ml/min;
[0057]
4、管路压力控制:氮气备压管路压力至20bar;
[0058]
5、将系统设置循环温度60℃,通过连续流反应器8块反应模块(204ml体积)反应300秒后,收集反应液。反应液中(2s,5r)-6-(羟基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺的纯度为96.89%。过滤脱除反应液中钯炭,转入磺化釜进行磺酸化(磺酸化试机为三氧化硫的络合物),经乙酸丁酯有机相萃取,离子对成盐(铵离子源为正三辛基甲基铵季铵盐),二氯甲烷萃取产物,无水硫酸钠干燥后,经减压浓缩,用4-甲基-2-戊酮进行套蒸析晶,过滤,使用4-甲基-2-戊酮淋洗,减压烘干得(2s,5r)-6-(磺基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺正三辛基甲基铵季铵盐。液相色谱纯度99.99%,最大单杂未检出,摩尔收率94.11%。
[0059]
对比例1:使用传统氢化高压反应釜,(2s,5r)-6-(磺基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺正四丁基季铵盐的制备
[0060]
1、装置:高压氢化反应器;
[0061]
2、料液配制:80.0g化合物(i)、2.4g10wt%钯炭、2.4g氢氧化钯催化剂、400ml水、400ml异丙醇,45.3g三氧化硫三甲胺,5.9g三乙胺,投入高压氢化反应釜中,进行氮气置换,氢气置换,搅拌。化合物(i)为:(2s,5r)-6-(苄基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺。
[0062]
3、间歇性通入氢气维持体系氢气压力1.0mpa;
[0063]
4、维持反应压力搅拌反应10小时;
[0064]
5、体系完成反应时间后,反应液中(2s,5r)-6-(羟基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺的纯度为88.89%,经过滤除钯,经乙酸丁酯有机相萃取,离子对成盐(铵离子源为正四丁基季铵盐正离子),二氯甲烷萃取产物,无水硫酸钠干燥后,经减压浓缩,用4-甲基-2-戊酮进行套蒸析晶,过滤,使用4-甲基-2-戊酮淋洗,减压烘干得(2s,5r)-6-(磺基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺正四丁基季铵盐。液相色谱纯度99.99%,最大单杂未检出,摩尔收率82.21%。
[0065]
对比例2:使用传统氢化高压反应釜,(2s,5r)-6-(磺基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺正三辛基甲基铵季铵盐的制备
[0066]
1、装置:高压氢化反应器;
[0067]
2、料液配制:80.0g化合物(i)、2.4g10wt%钯炭、2.4g氢氧化钯催化剂、400ml水、400ml异丙醇,45.3g三氧化硫三甲胺,5.9g三乙胺,投入高压氢化反应釜中,进行氮气置换,氢气置换,搅拌。化合物(i)为:(2s,5r)-6-(苄基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺。
[0068]
3、间歇性通入氢气维持体系氢气压力1.0mpa;
[0069]
4、维持反应压力搅拌反应1小时;
[0070]
5、体系完成反应时间后,反应液中(2s,5r)-6-(羟基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺的纯度为45.89%,经过滤除钯,经乙酸丁酯有机相萃取,离子对成盐(铵离子源为正三辛基甲基铵季铵盐正离子),二氯甲烷萃取产物,无水硫酸钠干燥后,经减压浓缩,用4-甲基-2-戊酮进行套蒸析晶,过滤,使用4-甲基-2-戊酮淋洗,减压烘干得
(2s,5r)-6-(磺基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺正三辛基甲基铵季铵盐。液相色谱纯度92.12%,最大单杂:7.12%,摩尔收率30.22%。
[0071]
对比例3:使用传统氢化高压反应釜,(2s,5r)-6-(磺基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺正三辛基甲基铵季铵盐的制备
[0072]
1、装置:高压氢化反应器;
[0073]
2、料液配制:80.0g化合物(i)、2.4g10wt%钯炭、2.4g氢氧化钯催化剂、400ml水、400ml异丙醇,45.3g三氧化硫三甲胺,5.9g三乙胺,投入高压氢化反应釜中,进行氮气置换,氢气置换,搅拌。化合物(i)为:(2s,5r)-6-(苄基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺。
[0074]
3、间歇性通入氢气维持体系氢气压力1.0mpa;
[0075]
4、维持反应压力搅拌反应10小时;
[0076]
5、体系完成反应时间后,反应液中(2s,5r)-6-(羟基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺的纯度为86.56%,经过滤除钯,经乙酸丁酯有机相萃取,离子对成盐(铵离子源为正三辛基甲基铵季铵盐正离子),二氯甲烷萃取产物,无水硫酸钠干燥后,经减压浓缩,用4-甲基-2-戊酮进行套蒸析晶,过滤,使用4-甲基-2-戊酮淋洗,减压烘干得(2s,5r)-6-(磺基氧基)-7-氧代-1,6-二氮杂二环[3.2.1]辛烷-2-甲酰胺正三辛基甲基铵季铵盐。液相色谱纯度99.80%,最大单杂:0.20%,摩尔收率81.01%。
[0077]
从上述实施例和对比例可知,实施例的产品(阿维巴坦钠中间体ii)摩尔收率在90%以上,产品纯度在99.0%以上,高于对比例的间歇式反应,而且反应时间大大缩短(实施例反应300s),而间歇式压力氢化反应釜反应的10小时,且其摩尔收率很低,间歇反应1小时的时候仅有30.22%。
[0078]
以上所述仅是对本发明的实施方式的举例展示,并非对本发明作任何形式上的限制。本发明保护范围以权利要求书为准且不由上述具体实施例所限,凡是依据本发明的技术实质对以上实施方式所作的任何简单修改或等同变化与修饰均属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1