制备邻氨基苯甲酸酯化合物的改进方法与流程
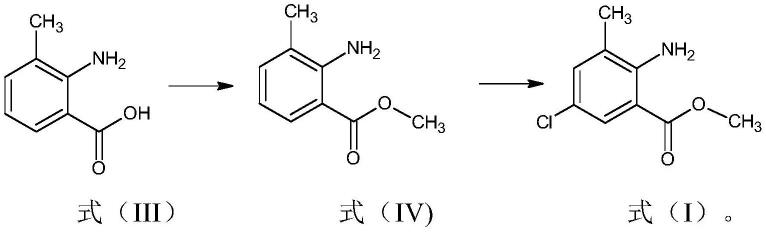
1.本发明涉及一种制备式(i)的邻氨基酸苯甲酸酯化合物的改进方法,
2.
背景技术:
3.邻氨基苯甲酸酯是制备许多医药和农产品的多用途且成本低廉的起始原料/中间体。
4.邻氨基苯甲酸酯已知可用于制备一类重要的合成杀虫剂,即二酰胺。二酰胺类杀虫剂分子具有相同的靶位点,即兰尼碱受体,并被归入杀虫剂抗性行动委员会(irac)作用模式分类的第28组,即兰尼碱受体调节剂。氯虫苯甲酰胺、氰虫苯甲酰胺、环苯甲酰胺、四氯虫苯甲酰胺和四苯甲酰胺等二酰胺类杀虫剂是众所周知的,且具有很强的效力。
5.现有技术已公开了多种制备二酰胺类杀虫剂的合成路线,许多方法都是通过式(i)的邻氨基苯甲酸酯化合物进行的。
[0006][0007]
制备中间体即式(i)的邻氨基苯甲酸酯的方法是本领域已知的。但是,现有技术中公开的反应顺序执行起来很繁琐,不适合商业化生产。这些方法中的一些牵涉使用昂贵的氯化剂,如次氯酸钠溶液、n-氯代琥珀酰亚胺等。大规模使用这样的昂贵试剂在经济上是不可行的。
[0008]
wo2006062978公开了一种通过在乙腈中使用硫酰氯氯化2-氨基-3-甲基苯甲酸甲酯来制备式(i)的邻氨基苯甲酸酯化合物的方法。所述方法包括在3-3.5小时内加入少量的硫酰氯,将温度保持在50-55℃,然后立即冷却至5℃,之后再进行后处理程序。指出了该方法的局限性,该方法要求非常缓慢地加入硫酰氯然后,再立即冷却至5℃。此外,反应所用的溶剂是乙腈,其具有吸湿性,可能导致反应混合物中水分含量的增加。众所周知,硫酰氯易与水反应,从而有可能形成硫氧化物形式的气态副产物,并且当反应进行时,这种气态副产物的形成可能会继续。在大规模生产中,气体逸出非常丰富,并且在任何不可预测的时间,
它会以突然激增的形式出现,导致类似反应失控的情况,从而导致反应物被抛出反应器。这些局限性和与该方法相关的问题使得该方法在商业规模上变得繁琐、低效和危险。
[0009]
因此,有必要找到一种在商业规模上进行该反应并克服与现有技术相关的一个或多个问题的安全方法。
技术实现要素:
[0010]
本发明的一个目的是提供一种制备式(i)的邻氨基苯甲酸酯化合物的方法。
[0011]
本发明的另一个目的是提供一种改进的、成本有效的且在工业上可行的制备式(i)的邻氨基苯甲酸酯化合物的方法。
[0012]
本发明的另一个目的是提供一种制备式(i)的邻氨基苯甲酸酯化合物的方法,该方法不要求极端温度条件和复杂的后处理步骤。
[0013]
根据本发明的一个方面,提供了一种在不分离式(iv)化合物的条件下制备式(i)化合物的方法。
[0014][0015]
根据本发明的另一个方面,提供了一种制备式(i)邻氨基酸甲酸酯化合物的方法,
[0016][0017]
该方法包括:
[0018]
a)用甲基化试剂将式(iii)化合物甲基化,获得式(iv)化合物,
[0019][0020]
b)用含氯溶剂提取式(iv)化合物;以及将分离的有机层用硫酰氯处理,获得式(i)的邻氨基苯甲酸酯化合物,
[0021][0022]
根据本发明的另一个方面,提供了一种制备式(i)的邻氨基苯甲酸酯化合物的方法,
[0023][0024]
该方法包括:
[0025]
a)用甲基化试剂将式(iii)化合物甲基化,获得式(iv)化合物,
[0026][0027]
b)用含氯溶剂处理式(iv)化合物,并将分离的有机层用硫酰氯处理,获得式(i)的邻氨基苯甲酸酯化合物,
[0028][0029]
其中,步骤a)中使用的式(iii)化合物通过还原式(ii)化合物而制备得到,
[0030][0031]
根据本发明的又一个方面,提供了一种制备式(i)的邻氨基甲酸酯化合物的方法,
[0032][0033]
该方法包括:
[0034]
a)用甲基化试剂将式(iii)化合物甲基化,获得式(iv)化合物,
[0035][0036]
b)用含氯溶剂提取式(iv)化合物,将分离的有机层用硫酰氯处理,获得式(i)的邻氨基苯甲酸酯化合物,
[0037][0038]
其中,步骤a)中使用的式(iii)化合物通过在铂催化剂存在的条件下还原式(ii)化合物而制备得到,
[0039]
具体实施方式
[0040]
提供以下描述以帮助全面理解本发明的示例性实施方案。它包括有助于理解的各种具体细节,但这些仅被视为是示例性的。本发明还包括本说明书中单独或共同提及或指示的所有这些步骤、特征、组合物和方法,以及任何两个或多个所述步骤或特征的任何和所有组合。
[0041]
定义
[0042]
为了方便起见,在进一步描述本发明之前,在此描述说明书中使用的某些术语、示例。这些定义应根据本公开的其余部分进行阅读,并由本领域技术人员理解。除非另有定义,否则本文中使用的所有技术和科学术语具有本领域普通技术人员通常理解的相同含义。本说明书中使用的术语定义如下,除非在特定情况下另有限制。
[0043]
除非另有说明,否则术语“室温”基本上意指20-35℃范围内的温度。
[0044]
术语“纯度”意指由hplc(“高效液相色谱”)测定的纯度。
[0045]
如本文所用,术语“大约”或“近似”包含所述值,并且考虑到所述测量和与特定量的测量相关的误差(即,测量系统的限制),意指在由本领域普通技术人员确定的特定值的可接受偏差范围内。例如,“大约”可意指在一个或多个标准偏差范围内,或在所述值的
±
10或
±
5范围内。除非本文中另有说明,否则数值范围的列举仅旨在作为单独引用落入该范围内的每个单独数值的简写方法,并且每个单独数值被并入本说明书中,如同其在本文中单独列举一样。所有范围的端点都包含在该范围内,并可独立组合。应当理解,在提供参数范围的条件下,还提供该范围内的所有整数及其十分之一。例如,“0.1-80%”包括0.1%、0.2%、0.3%等,最高可达80%。如本文所用,术语“包含”、“包括”、“具有”、“含有”、“涉及”等应理解为是开放式的,即意指包括但不限于。
[0046]
术语“优选的”和“优选地”是指在某些情况下可能提供某些益处的本发明实施方案。在一个实施方案中,本文所描述的方面和实施方案也应解释为将表述“包括”替换为“由
…
组成”或“基本上由
…
组成”或“大体上由
…
组成”。
[0047]
本发明描述了一种制备式(i)的邻氨基苯甲酸酯化合物的改进方法。本发明的方法基于新的反应顺序,该反应顺序可容易地在大批量生产中实施,以高产量地提供所需的化合物,但不涉及释放因使用硫酰氯而出现的剧烈的气态副产物的任何风险。因此,本发明提供的方法经济、环保、安全。
[0048]
根据本发明的一个方面,提供了一种在不需要分离式(iv)化合物的条件下制备式(i)的邻氨基苯甲酸酯化合物的方法,
[0049][0050]
在一个实施方案中,提供了一种制备式(i)的邻氨基苯甲酸酯的方法,
[0051][0052]
该方法包括:
[0053]
i)用甲基化试剂将式(iii)化合物甲基化,获得式(iv)化合物;以及
[0054]
ii)用硫酰氯处理式(iv)化合物,获得式(i)的邻氨基苯甲酸酯,
[0055][0056]
其中,所述方法在不分离式(iv)化合物的条件下进行。
[0057]
在一个实施方案中,所用的甲基化试剂选自碘甲烷、碳酸二甲酯、硫酸二甲酯、重氮甲烷、二甲氧基丙烷、二甲基锌、氟磺酸甲酯、三氟甲磺酸甲酯等,优选使用硫酸二甲酯。
[0058]
相对于式(iii)化合物使用的甲基化试剂的量为1至3摩尔。
[0059]
在一个实施方案中,所述甲基化在溶剂存在的条件下进行。
[0060]
所用的溶剂选自有机溶剂,如二氯甲烷、二氯乙烷、乙腈、二甲基甲酰胺、二甲基乙酰胺、n-甲基吡咯烷酮及其混合物。
[0061]
在一个实施方案中,甲基化反应在二甲基甲酰胺作为溶剂的存在的条件下进行。
[0062]
在一个实施方案中,甲基化反应在二甲基乙酰胺作为溶剂的存在的条件下进行。
[0063]
相对于式(iii)化合物使用的溶剂的量约为4至10倍。
[0064]
在一个实施方案中,甲基化反应在碱存在的条件下进行。
[0065]
通常,使用的碱选自氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾等。
[0066]
相对于式(iii)化合物使用的碱的量为1至1.5摩尔。
[0067]
在一个实施方案中,甲基化反应在20至100℃范围内的温度下进行。
[0068]
在一个实施方案中,甲基化反应进行0.5至6小时。
[0069]
在一个实施方案中,所述方法的步骤i)还包括用含氯溶剂分离式(iv)化合物,并且降低有机层的水分含量至小于0.5%w/w。
[0070]
在一个实施方案中,所用的含氯溶剂选自由二氯甲烷、二氯乙烷、氯仿、1,1,2-三氯乙烷、氯苯、邻二氯苯等组成的组。
[0071]
在一个实施方案中,所用的含氯溶剂为二氯乙烷。
[0072]
相对于式(iii)化合物使用的含氯溶剂的量约为3至10倍。
[0073]
在一个实施方案中,将有机层的水分含量降低至小于0.5%w/w。
[0074]
在一个实施方案中,步骤i)中得到的式(iv)化合物不被分离,并将步骤i)中得到的有机层直接用于步骤ii)中以进行进一步处理。
[0075]
在一个实施方案中,可通过蒸馏、沉淀等常规已知的方法分离步骤i)中得到的式(iv)化合物,然后直接用于步骤ii)中。
[0076]
在一个实施方案中,步骤ii)中,相对于式(iii)化合物使用的硫酰氯的量为0.5摩尔至1.5摩尔。
[0077]
在一个实施方案中,硫酰氯处理在20℃至100℃的温度下进行。在一个实施方案中,硫酰氯处理在40℃至70℃的温度下进行。
[0078]
在一个实施方案中,用硫酰氯进行处理10分钟至3小时。
[0079]
根据本发明的另一个方面,提供了一种制备式(i)的邻氨基苯甲酸酯化合的方法,
[0080][0081]
该方法包括:
[0082]
i)用甲基化试剂将式(iii)化合物甲基化,获得式(iv)化合物,
[0083][0084]
ii)通过用含氯有机溶剂进行处理来分离式(iv)化合物;以及将分离的有机层用硫酰氯进一步处理,获得式(i)的邻氨基苯甲酸酯化合物,
[0085][0086]
在一个实施方案中,所用的含氯溶剂选自二氯甲烷、二氯乙烷、氯仿、1,1,2-三氯乙烷、氯苯、邻二氯苯等。
[0087]
在一个实施方案中,所用的含氯溶剂为二氯乙烷。
[0088]
相对于式(iii)化合物使用的含氯溶剂的量约为3至10倍。
[0089]
在一个实施方案中,所述方法的步骤ii)还包括在用硫酰氯进行处理之前将有机层的水分含量降低至小于0.5%w/w。
[0090]
在一个实施方案中,通过共沸蒸馏降低有机层的水分含量。
[0091]
在一个实施方案中,将水分含量降低至小于0.5%w/w。
[0092]
优选地,将有机层的水分含量调节至小于0.1%w/w。
[0093]
在一个实施方案中,相对于式(iii)化合物使用的硫酰氯的量为0.5至1.5摩尔。
[0094]
在一个实施方案中,在催化剂存在的条件下进行步骤ii)。优选地,所用的催化剂为二甲基甲酰胺。
[0095]
在一个实施方案中,硫酰氯处理在20℃至100℃的温度下进行。在一个实施方案中,硫酰氯处理在40℃至70℃的温度下进行。
[0096]
在一个实施方案中,用硫酰氯进行处理10分钟至3小时。
[0097]
本发明人通过仔细选择本发明方法中使用的溶剂已巧妙地设计了本发明方法,所述溶剂在本质上不具有吸湿性。使用这种溶剂可以在很大程度上调节水分含量。
[0098]
此外,通过使用含氯有机溶剂提取式(iv)化合物,也可以避免在所述方法中使用多种溶剂。通常,可以将调节水分含量后的有机层直接用于氯化反应,而无需分离式(iv)化合物,从而使所述方法高效、环保和经济地用于大规模生产。
[0099]
同样,由于不需要分离式(iv)化合物,因此减少了因结晶或沉淀或过滤等固体处理步骤而导致的产率损失的可能性。
[0100]
根据本发明的一个实施方案,通过还原式(ii)化合物来制备式(iii)化合物,
[0101][0102]
在一个实施方案中,将式(ii)化合物还原为式(iii)化合物的步骤在催化剂和溶剂存在的条件下进行。
[0103]
通常,所用的催化剂选自钯催化剂、铂催化剂或雷尼镍。可以使用钯或铂催化剂,如碳载钯或碳载铂。优选地,使用铂催化剂。
[0104]
在一个实施方案中,在将式(ii)化合物还原为式(iii)化合物的步骤中使用的铂催化剂可以再循环和重复用于相同反应20次以上,从而使所述方法具有成本效益且在工业规模上经济上可行。
[0105]
在一个实施方案中,在将式(ii)化合物还原为式(iii)化合物的步骤中使用的溶剂选自腈类(例如,乙腈)或c1-c5醇(例如,甲醇、乙醇、异丙醇、正丙醇、丁醇、叔丁醇)等。
[0106]
在一个实施方案中,将式(ii)化合物还原为式(iii)化合物的步骤在20至80℃的温度下进行。
[0107]
在一个实施方案中,将式(ii)化合物还原为式(iii)化合物的步骤进行1至10小时。
[0108]
根据本发明的另一个方面,提供了一种制备式(i)的邻氨基苯甲酸酯化合物的方法,
[0109][0110]
该方法包括:
[0111]
i)用甲基化试剂将式(iii)化合物甲基化,获得式(iv)化合物,
[0112][0113]
ii)在含氯有机溶剂中提取式(iv)化合物,并将分离的有机层用硫酰氯处理,获得式(i)的邻氨基苯甲酸酯化合物,
[0114][0115]
其中,通过还原式(ii)化合物来制备步骤i)中使用的式(iii)化合物,
[0116][0117]
根据本发明的又一个方面,提供了一种制备式(i)的邻氨基苯甲酸酯化合物的方法,
[0118][0119]
该方法包括:
[0120]
i)用甲基化试剂将式(iii)化合物甲基化,获得式(iv)化合物,
[0121][0122]
ii)用含氯有机溶剂提取式(iv)化合物,并将分离的有机层用硫酰氯处理,获得式(i)的邻氨基苯甲酸酯化合物,
[0123][0124]
其中,通过在铂催化剂存在的条件下还原式(ii)化合物来制备步骤i)中使用的式(iii)化合物,
[0125][0126]
根据本发明的一个方面,提供了一种制备式(i)的邻氨基苯甲酸酯化合物的方法,
[0127][0128]
该方法包括:
[0129]
i)用甲基化试剂将式(iii)化合物甲基化,获得式(iv)化合物,
[0130][0131]
ii)在含氯有机溶剂中提取式(iv)化合物,将有机层中的水分含量降低至小于0.5%w/w;
[0132]
iii)用硫酰氯处理有机层,获得式(i)的邻氨基苯甲酸酯化合物,
[0133][0134]
其中,通过还原式(ii)化合物来制备步骤i)中使用的式(iii)化合物,
[0135][0136]
根据本发明的又一个方面,提供了一种制备式(i)的邻氨基苯甲酸酯化合物的方法,
[0137][0138]
该方法包括:
[0139]
i)用甲基化试剂将式(iii)化合物甲基化,获得式(iv)的酯,
[0140][0141]
ii)在含氯溶剂中提取式(iv)的酯,获得有机层;
[0142]
iii)将有机层的水分含量降低至小于0.5%w/w;
[0143]
iv)用硫酰氯处理步骤c)中的有机层,获得式(i)的邻氨基苯甲酸酯化合物,
[0144][0145]
其中,通过在铂催化剂存在的条件下还原式(ii)化合物来制备式(iii)化合物,
[0146][0147]
在一个实施方案中,可以使用式(i)化合物来制备二酰胺类杀虫剂化合物,例如氯虫苯甲酰胺、氰虫苯甲酰胺、环苯甲酰胺、四氯虫苯甲酰胺和四苯甲酰胺。优选地,式(i)化合物用于制备氯虫苯甲酰胺。
[0148]
本发明具有如下优点:1.所述方法是一种制备式(i)的邻氨基苯甲酸酯化合物的简单、成本有效且工业上可行的方法。2.所述方法不需要分离式(iv)化合物,从而减少了流出物的生成和操作步骤。3.涉及使用成本有效的试剂,如硫酰氯。4.用于还原式(ii)化合物的铂催化剂可循环使用多次。
[0149]
实施例
[0150]
通过下面例举的实验确定本发明所述的制备式(i)的邻氨基苯甲酸酯化合物的方法。这些实施例仅仅是说明性的,不应理解为以任何方式限制本发明的范围和基本原理。实际上,除了本文所示和描述的那些以外,根据以下实施例和前述描述,本发明的各种修改对本领域的技术人员来说是显而易见的。
[0151]
分析方法详情:
[0152]
用cosmosil-5c18-ms-ii柱在配备uv检测器的高效液相色谱仪上分析样本。
[0153]
实施例1:2-硝基-3-甲基苯甲酸-式(ii)化合物的制备
[0154]
将6147g发烟硝酸装入反应容器中并冷却至-15至-10℃,然后加入1639g间甲苯甲酸,将反应混合物保持在相同温度下2小时。反应完成后,分离粗2-硝基-3-甲基苯甲酸,加入1180g 50%乙醇。将混合物加热至75-80℃,持续1小时,然后冷却至室温,产物过滤并干燥,获得1000g的2-硝基-3-甲基苯甲酸。
[0155]
实施例2:2-氨基-3-甲基苯甲酸-式(iii)化合物的制备
[0156]
将1032g 2-硝基-3-甲基苯甲酸、51g的3重量%活性碳载铂(pt/c)催化剂和6640g甲醇加入至反应容器中。将混合物在30℃下以4-6kg/cm2进行氢化,然后将反应混合物的温度升至50-55℃,并保持在该温度6小时。反应完成后,过滤反应混合物以回收催化剂,蒸馏滤液以回收大约70%所使用的溶剂并获得反应物质。然后,加入1674g水,将混合物在20-25℃保持1小时以使产物沉淀。产物过滤并干燥,获得806g 2-氨基-3-甲基苯甲酸。
[0157]
实施例3:3-甲基-2-氨基苯甲酸甲酯-式(iv)化合物的制备
[0158]
向5250g二甲基甲酰胺与806g 2-氨基-3-甲基苯甲酸的混合物中加入850g碳酸钾,将反应混合物加热至50-55℃。保持温度到50-55℃,在2-3小时内加入1380g硫酸二甲酯,升温至65-70℃。将混合物保持在该温度下3至4小时。反应完成后,蒸馏出所使用的溶剂,将4050g二氯乙烷加入至由此而获得的残留物中。然后向混合物中加入4850g水,在室温下搅拌混合物1小时。1小时后,分层,在550-650mmhg真空和50-55℃的条件下共沸蒸馏有机层。将有机层的水分含量降低至0.1%以下,并且在实施例4中使用该包含3-甲基-2-氨基苯
甲酸酯的有机层。
[0159]
实施例4:3-甲基-2-氨基-5-氯苯甲酸甲酯-式(i)化合物的制备
[0160]
在50-55℃下、2至3小时内向在实施例3获得的有机层中加入660mg硫酰氯,并保持反应1小时。反应完成后,将混合物冷却至室温,加入1200g水,然后加入920g氢氧化钠以将ph调节至5.5至6之间。分层,蒸馏出有机层,获得1000g 3-甲基-2-氨基-5-氯苯甲酸甲酯。
[0161]
实施例5:3-甲基-2-氨基-5-氯苯甲酸甲酯-式(i)化合物的制备
[0162]
步骤1:2-硝基-3-甲基苯甲酸的制备
[0163]
将735g发烟硝酸装入反应容器中并冷却至-15至-10℃。然后逐步加入250g间甲苯甲酸,将反应混合物保持在相同温度下2小时。反应完成后,分离粗2-硝基-3-甲基苯甲酸,向其中加入225g 50%乙醇。将混合物加热至75-80℃保持1小时,然后冷却至室温,产物过滤并干燥,获得161g 2-硝基-3-甲基苯甲酸。
[0164]
步骤2:2-氨基-3-甲基苯甲酸的制备
[0165]
将159g 2-硝基-3-甲基苯甲酸、7g的3重量%活性碳载铂(pt/c)催化剂和1000g甲醇加入至反应容器中。将混合物在30℃下以4-6kg/cm2进行氢化,然后将反应混合物的温度升至50-55℃,并在该温度保持6小时。反应完成后,过滤反应混合物以回收催化剂,蒸馏滤液以回收大约70%所使用的溶剂并获得反应物质。向反应物质中加入525g水,将混合物在20-25℃保持1小时以使产物沉淀。产物过滤并干燥,获得120g 2-氨基-3-甲基苯甲酸。
[0166]
步骤3:3-甲基-2-氨基苯甲酸甲酯的制备
[0167]
向650g二甲基甲酰胺与步骤2中的100g 2-氨基-3-甲基苯甲酸的混合物中加入106g碳酸钾,将获得的反应混合物加热至50-55℃。将温度保持在50-55℃,在2-3小时内加入170g硫酸二甲酯,升温至65-70℃。将混合物在该温度保持3至4小时。反应完成后,蒸馏出所使用的溶剂,并加入600g水。然后向混合物中加入500g二氯乙烷,室温下搅拌混合物1小时。1小时后,分层,在550-650mmhg真空和50-55℃的条件下共沸蒸馏有机层。将有机层的水分含量降低至0.1%以下,并且在步骤4中使用该包含3-甲基-2-氨基苯甲酸酯的有机层。
[0168]
步骤4:3-甲基-2-氨基5-氯苯甲酸甲酯的制备
[0169]
在50-55℃下、2至3小时内向在步骤3获得的有机层中加入82g硫酰氯,并保持反应1小时。反应完成后,将混合物冷却至室温,加入150g水,然后加入114g氢氧化钠以将ph调节至5.5至6。分层,蒸馏出有机层,获得124g 3-甲基-2-氨基-5-氯苯甲酸甲酯。
[0170]
实施例6:3-甲基-2-氨基-5-氯苯甲酸甲酯-式(i)化合物的制备步骤1和步骤2以与实施例5相似的方式进行。
[0171]
步骤3:3-甲基-2-氨基苯甲酸甲酯的制备
[0172]
向650g二甲基甲酰胺与100g 2-氨基-3-甲基苯甲酸的混合物中加入106g碳酸钾,将获得的反应混合物加热至50-55℃。在2-3小时内向混合物中加入170g硫酸二甲酯,升温至65-70℃。将混合物在该温度保持3至4小时。反应完成后,蒸馏出所使用的溶剂,然后加入580g二氯乙烷和580g水。混合物搅拌并加热至40-45℃的温度。分层,在550-650mmhg真空和50-55℃温度的条件下共沸蒸馏有机层。将有机层的水分含量降低至0.1%以下,并且在步骤4中使用该包含3-甲基-2-氨基苯甲酸酯的有机层。
[0173]
步骤4:3-甲基-2-氨基5-氯苯甲酸甲酯的制备
[0174]
在50-55℃下、2至3小时内向在步骤3获得的有机层中加入82g硫酰氯和1g二甲基
甲酰胺。然后将反应混合物保持在相同温度下1-2小时。反应完成后,将混合物冷却至室温,加入150g水,然后加入50%氢氧化钠水溶液以将ph调节至5.5至6。分层,蒸馏出有机层,获得124g hplc纯度为99.21%的3-甲基-2-氨基-5-氯苯甲酸甲酯。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1