一种4-胺基酚类衍生物及应用
1.本发明涉及化学生物学技术领域,具体涉及4-胺基酚类衍生物及应用。
背景技术:
2.酪氨酸酶(tyrosinase,tyr)是人体内一类介导黑色素合成的代谢酶,与多种疾病的发病过程有关。该酶的功能减退或缺失时,会导致白癜风或白化病等色素合成障碍性皮肤病;相反,若该酶的表达量过多,又可因黑色素生成旺盛而出现雀斑、暗沉、黄褐斑、老年斑等色素沉着症状。随着研究的深入,人们发现有些复杂结构的酪氨酸酶抑制剂可同时作为抗菌试剂使用,这进一步丰富了化合物功能,使得此类化合物在医美赛道有极大的潜力,但是此类化合物通常为巴比妥类、吡唑啉类、芦荟大黄素类等具有复杂结构化合物。
3.对胺基酚衍生物因其多种生物活性而受到广泛关注,该骨架被视为两性电解质,具有良好的水溶性,并且这些骨架能够将酚羟基的氢原子提供给自由基在氧化过程中阻止链传播,因此此类化合物具有很好的生物活性。
4.然而,国内外研究都关注于脂肪仲烷胺基酚类化合物,由于烷基供电子效应导致化合物电子云密度较高而缺乏稳定性,同时特殊的仲烷胺基酚结构也是导致其衍生性差的原因之一,因而国内外对该结构的进一步生物活性研究较少。
5.目前,对胺基酚衍生物主要作为着色剂而被研究开发,但是对于兼具酪氨酸酶活性酸酶抑制活性、抗氧化和抗菌活性的化合物的开发研究还未见报道。因此,本发明通过引入n-芳基使胺基酚结构稳定的同时,增强化合物可衍生性,最终从衍生化合物中成功找到了若干同时具有良好氨酸酶抑制活性、抗氧化和抗菌活性的化合物。
技术实现要素:
6.为解决上述技术问题,本发明的目的在于提供一种4-胺基酚类衍生物及应用。通过引入n-芳基使胺基酚结构稳定的同时,增强化合物可衍生性,最终从衍生化合物中成功找到了若干同时具有良好氨酸酶抑制活性、抗氧化和抗菌活性的化合物。
7.为实现上述目的,本发明的技术方案如下。
8.一种4-胺基酚类衍生物,其结构通式如下式(
ⅰ‑
1)或式(
ⅰ‑
2)所示:
[0009][0010]
式(
ⅰ‑
1)和(
ⅰ‑
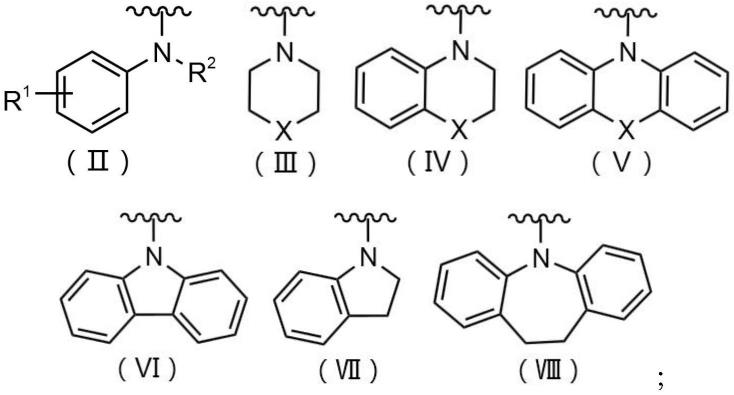
2)中,d1、d2、d3分别独立选自式(ⅱ)、式(ⅲ)、式(ⅳ)、式(
ⅴ
)、式(ⅵ)、式(ⅶ)或式(
ⅷ
)所示基团:
[0011][0012]
式(ⅱ)中,r1选自-h、-f、-cl、-br、c1~c4的烷基、硝基、氰基、甲氧基、取代或未被取代的芳基、取代或未被取代的稠环芳基、取代或未被取代的稠杂环、取代或未被取代的芳基乙炔基中的任意一种;
[0013]
r2选自c1~c14的烷基、烯丙基、取代或未被取代的单杂环烷基、取代或未被取代的稠杂环烷基、取代或未被取代的芳基、取代或未被取代的稠环芳基、取代或未被取代的芳基烷基、取代或未被取代的稠环芳基烷基、取代或未被取代的芳氨基烷基中的任意一种;
[0014]
式(ⅲ)~式(
ⅴ
)中,x为氧原子、硫原子、碳原子中的任意一种。
[0015]
进一步,式(ⅱ)中,当r1选自取代的芳基、取代的稠环芳基、取代的稠杂环、取代的芳基乙炔基中的任意一种时,取代基为-f、-cl、-br、c1~c4的烷基、硝基、氰基、甲氧基中的任意一种;
[0016]
当r2选自取代的单杂环烷基、取代的稠杂环烷基、取代的芳基、取代的稠环芳基、取代的芳基烷基、取代的稠环芳基烷基、取代的芳氨基烷基中的任意一种时,取代基为-f、-cl、-br、c1~c4的烷基、硝基、氰基、甲氧基中的任意一种。
[0017]
更进一步,式(ⅱ)所代表的基团选自以下结构式中的一种:
[0018][0019][0020]
更进一步,式(ⅲ)所代表的基团选自以下结构式中的一种:
[0021]
进一步,具体为如下化合物中的一种:
[0022]
[0023][0024]
本发明还提供一种4-胺基酚类衍生物在制备酪氨酸酶抑制剂中的应用。
[0025]
本发明还提供一种4-胺基酚类衍生物在制备针对mrsa(耐甲氧西林金黄色葡萄球菌)的抗菌剂中的应用。
[0026]
本发明还提供一种4-胺基酚类衍生物在制备抗氧化剂中的应用。
[0027]
本发明的有益效果:
[0028]
1、本发明通过引入n-芳基使胺基酚结构稳定的同时,增强化合物可衍生性,最终从衍生化合物中成功找到了若干同时具有良好氨酸酶抑制活性、抗氧化和抗菌活性的化合物。
[0029]
2、针对n-单烷基取代的对胺基苯酚通常不稳定易被氧化及衍生结构单一的问题,本发明通过引入n-芳基对胺基酚使结构稳定,增强化合物可衍生性,对此类结构的抗菌、抗氧化和酪氨酸抑制活性进行评价,找到了若干同时具有酪氨酸酶抑制活性和抗菌活性的化合物。本发明具有化合物合成简单,针对mrsa(耐甲氧西林金黄色葡萄球菌)抑菌效果好,酶抑制率高等特点,有着广阔的应用前景。
附图说明
[0030]
图1是不同浓度化合物4k,4m,4n,4p,4ab的对raw 264.7细胞的毒性测试柱形图。
[0031]
图2是化合物对酪氨酸酶的影响结果。其中,(a)是各种化合物在16μm时对酪氨酸酶活性的影响。(b)是化合物对蘑菇酪氨酸酶活性的浓度依赖效应(平均值)。
[0032]
图3是在化合物4ab存在下酪氨酸酶氧化l-酪氨酸的动力学图。其中,(a)是在0、1、2、4、8、16和32μm浓度下,3-1j抑制酪氨酸酶的lineweaver-burk图。(b)是斜率与抑制剂(3-1j)浓度的曲线图。
[0033]
图4是不同浓度抗坏血酸、4k,4m,4n,4p和4ab对dpph的清除作用柱形图。
具体实施方式
[0034]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0035]
基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0036]
cu(otf)2,三氟甲烷磺酸铜(ⅱ)。schlenk管,舒仑克管。thf,四氢呋喃。buchwald
–
hartwig偶联反应,布赫瓦尔德-哈特维希反应。pd2(dba)3,三(二亚苄基丙酮)二钯。davephos,2-二环己膦基-2'-(n,n-二甲胺)-联苯。lin(tms)2,双-(三甲基硅基)胺锂。ruphos,2-双环已基膦-2',6'-二异丙氧基联苯。下述各实施例中所述实验方法如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可在市场上购买得到。
[0037]
下面我们对不同类型的化合物为例,提供这些化合物的合成方法。
[0038]
实施例1
[0039]
n-芳基对胺基酚类衍生物的合成方法,该方法是以环己烯酮及n-烷基苯胺为底物,以cu(otf)2为催化剂,l为配体,甲苯为溶剂,氧气气氛中于80℃条件下反应8-60h,得到目标产物n-芳基对胺基酚类衍生物,其反应过程如下所示:
[0040][0041]
具体操作如下:将带有磁子的schlenk管除水,依次加入mgso4(3.0eq.,干燥剂),cu(otf)2(5.0mol%)和l(10.0mol%),然后排空并用氧气置换(三次)。用针管依次向反应管中加入环己烯酮(3.0eq.),n-烷基苯胺(0.3mmol,1.0eq.)和三氟乙酸(2.5mol%,预先配置成三氟乙酸-甲苯溶液,现制现用),根据不同底物在80℃条件下反应8-60h。待反应完全后,过滤不容物,用乙酸乙酯洗涤三次,浓缩后柱层析,得到目标产物n-芳基对胺基酚类衍生物。化合物1~27以及化合物40~58均采用实施例1的方法制备。
[0042]
实施例2
[0043]
n-芳基对环烷胺基酚类衍生物的合成方法,该方法是以对溴苯酚及二级胺为底物,以pd2(dba)3为催化剂,davephos(2-二环己膦基-2'-(n,n-二甲胺)-联苯)为配体,thf为溶剂,氮气气氛中于65℃条件下反应8-12h,得到目标产物n-芳基对环烷胺基酚类衍生物,其反应过程如下所示:
[0044][0045]
将pd2(dba)3(2.0mol%pd),davephos(2-二环己膦基-2'-(n,n-二甲胺)-联苯)(2.4mol%),对溴苯酚(5.0mmol)及二级胺(6.0mmol)依次加入到除水的schlenk瓶中,氮气置换三次后于冰水浴中用针管缓慢加入lin(tms)2(1m,lin(tms)
2-thf溶液,5.5ml),在65℃下反应8-12h后冷至室温,加入1m hcl(5-10ml)淬灭,用饱和nahco3调至碱性,用乙酸乙酯萃取三次合并有机相,干燥后浓缩,硅胶柱层析,洗脱剂为乙酸乙酯:石油醚=1:20到1:8。化合物32~34采用实施例2的方法制备。
[0046]
实施例3
[0047]
n-芳基对苯并环烷胺基酚类衍生物的合成方法,该方法是以对溴苯甲醚及二级胺为底物,以pd(oac)2为催化剂,ruphos为配体,dcm/h2o为溶剂,氮气气氛中于110℃条件下反应8-12h,得到目标产物n-芳基对苯并环烷胺基酚类衍生物,其反应过程如下所示:
[0048][0049]
在除水的圆底烧瓶中加入对溴苯甲醚(5.1mmol),二级胺(5.0mmol),pd(oac)2(0.05mmol),ruphos(0.10mmol)及nao
t
bu(6.0mmol),氮气置换三次,于110℃下反应8-12h,冷至室温后加入dcm/h2o=1:1混合液。萃取有机相三次后干燥浓缩,柱层析,洗脱剂为二氯甲烷/甲叔醚。
[0050]
将得到的对胺基醚类化合物溶于除水的dcm(30ml)后加入到100ml圆底烧瓶中(2.2mmol),在-78℃下缓慢滴加1m的bbr3(3ml,3.0mmol),室温反应3h后,将混合液倒入到冰水中,缓慢滴加饱和nahco3直至溶液显碱性,dcm萃取后干燥,浓缩,硅胶柱层析,洗脱剂为乙酸乙酯:石油醚=1:20到1:8。化合物28、30采用实施例3的方法制备。
[0051]
实施例4
[0052]
n-芳基对稠杂环烷胺基酚类衍生物的合成方法,该方法是以对溴苯甲醚及二级胺为底物,以pd(oac)2为催化剂,ruphos为配体,dcm/h2o为溶剂,氮气气氛中于110℃条件下反应8-12h,得到目标产物n-芳基对稠杂环烷胺基酚类衍生物,其反应过程如下所示:
[0053][0054]
在除水的圆底烧瓶中加入对溴苯甲醚(5.1mmol),二级胺(5.0mmol),pd(oac)2(0.05mmol),ruphos(0.10mmol)及nao
t
bu(6.0mmol),氮气置换三次,于110℃下反应8-12h,冷至室温后加入dcm/h2o=1:1混合液。萃取有机相三次后干燥浓缩,柱层析,洗脱剂为二氯甲烷/甲叔醚。
[0055]
将得到的对胺基醚类化合物溶于除水的dcm(30ml)后加入到100ml圆底烧瓶中
(2.2mmol),在-78℃下缓慢滴加1m的bbr3(3ml,3.0mmol),室温反应3h后,将混合液倒入到冰水中,缓慢滴加饱和nahco3直至溶液显碱性,dcm萃取后干燥,浓缩,硅胶柱层析,洗脱剂为乙酸乙酯:石油醚=1:20到1:8。化合物29、59、61采用实施例4的方法制备。
[0056]
实施例5
[0057]
n-芳基对稠杂环烷胺基酚类衍生物的合成方法,该方法是以对溴苯甲醚及二级胺为底物,以pd(oac)2为催化剂,ruphos为配体,dcm/h2o为溶剂,氮气气氛中于110℃条件下反应8-12h,得到目标产物n-芳基对稠杂环烷胺基酚类衍生物,其反应过程如下所示:
[0058][0059]
在除水的圆底烧瓶中加入对溴苯甲醚(5.1mmol),二级胺(5.0mmol),pd(oac)2(0.05mmol),ruphos(0.10mmol)及nao
t
bu(6.0mmol),氮气置换三次,于110℃下反应8-12h,冷至室温后加入dcm/h2o=1:1混合液。萃取有机相三次后干燥浓缩,柱层析,洗脱剂为二氯甲烷/甲叔醚。
[0060]
将得到的对胺基醚类化合物溶于除水的dcm(30ml)后加入到100ml圆底烧瓶中(2.2mmol),在-78℃下缓慢滴加1m的bbr3(3ml,3.0mmol),室温反应3h后,将混合液倒入到冰水中,缓慢滴加饱和nahco3直至溶液显碱性,dcm萃取后干燥,浓缩,硅胶柱层析,洗脱剂为乙酸乙酯:石油醚=1:20到1:8。化合物60采用实施例5的方法制备。
[0061]
实施例6
[0062]
n-芳基对稠杂环烷胺基酚类衍生物的合成方法,该方法是以对溴苯甲醚及二级胺为底物,以pd(oac)2为催化剂,ruphos为配体,dcm/h2o为溶剂,氮气气氛中于110℃条件下反应8-12h,得到目标产物n-芳基对稠杂环烷胺基酚类衍生物,其反应过程如下所示:
[0063][0064]
在除水的圆底烧瓶中加入对溴苯甲醚(5.1mmol),二级胺(5.0mmol),pd(oac)2(0.05mmol),ruphos(0.10mmol)及nao
t
bu(6.0mmol),氮气置换三次,于110℃下反应8-12h,冷至室温后加入dcm/h2o=1:1混合液。萃取有机相三次后干燥浓缩,柱层析,洗脱剂为二氯甲烷/甲叔醚。
[0065]
将得到的对胺基醚类化合物溶于除水的dcm(30ml)后加入到100ml圆底烧瓶中(2.2mmol),在-78℃下缓慢滴加1m的bbr3(3ml,3.0mmol),室温反应3h后,将混合液倒入到冰水中,缓慢滴加饱和nahco3直至溶液显碱性,dcm萃取后干燥,浓缩,硅胶柱层析,洗脱剂为乙酸乙酯:石油醚=1:20到1:8。化合物31采用实施例6的方法制备。
[0066]
实施例7
[0067]
n-芳基对稠杂环烷胺基酚类衍生物的合成方法,该方法是以对溴苯甲醚及二级胺为底物,以pd(oac)2为催化剂,ruphos为配体,dcm/h2o为溶剂,氮气气氛中于110℃条件下反应8-12h,得到目标产物n-芳基对稠杂环烷胺基酚类衍生物,其反应过程如下所示:
[0068][0069]
在除水的圆底烧瓶中加入对溴苯甲醚(5.1mmol),二级胺(5.0mmol),pd(oac)2(0.05mmol),ruphos(0.10mmol)及nao
t
bu(6.0mmol),氮气置换三次,于110℃下反应8-12h,冷至室温后加入dcm/h2o=1:1混合液。萃取有机相三次后干燥浓缩,柱层析,洗脱剂为二氯甲烷/甲叔醚。
[0070]
将得到的对胺基醚类化合物溶于除水的dcm(30ml)后加入到100ml圆底烧瓶中(2.2mmol),在-78℃下缓慢滴加1m的bbr3(3ml,3.0mmol),室温反应3h后,将混合液倒入到冰水中,缓慢滴加饱和nahco3直至溶液显碱性,dcm萃取后干燥,浓缩,硅胶柱层析,洗脱剂为乙酸乙酯:石油醚=1:20到1:8。化合物62采用实施例7的方法制备。
[0071]
实施例8
[0072]
2,4-二胺基酚类衍生物的合成方法,该方法是以n-甲基苯胺及对胺基苯酚类化合物为底物,以cu(otf)2为催化剂,ti(oipr)4为添加剂,甲苯为溶剂,于室温下反应7-10h,得到目标产物2,4-二胺基酚类衍生物,其反应过程如下所示。
[0073][0074]
在除水的反应管中加入磁子,na2so4(85mg,2.0eq.,0.6mmol)和cu(otf)2(16mg,0.045mmol,15mol%),将n-甲基苯胺(35mg,0.33mmol,1.1eq.),对胺基苯酚类化合物(0.3mmol,1.0eq.)溶于3ml除水甲苯后加入反应管,封口,用针管缓慢加入ti(oipr)4(25.0mg,30mol%),室温下反应7-10h,待反应结束后用0.1m hcl(5ml)淬灭,饱和碳酸氢钠萃取ph》8,无水硫酸钠干燥,硅胶柱层析分离。化合物35~39采用实施例8的方法制备。
[0075]
采用实施例1~8的方法制备的系列化合物的收率和核磁波谱数据如下:
[0076]
化合物1(4a):实施例1方法,蓝黑色油状液体,73%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ=7.15
–
7.08(m,2h,ph),6.96(d,j=8.8hz,2h,ph),6.79(d,j=8.8hz,2h,ph),6.74
–
6.66(m,3h,ph),3.17(d,j=10.4hz,3h,-ch3)。
[0077]
化合物2(4b):实施例1方法,黑色油状液体,79%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4):δ=7.08-7.05(m,2h,ph),6.94(d,j=10hz,2h,ph),6.79(d,j=5hz,2h,ph),6.65
–
6.61(m,3h,ph),3.64(q,j=6.7hz,2h,ch
2-n),1.14(t,j=7.5hz,3h,ch
3-ch2n)。
[0078]
化合物3(4c):实施例1方法,黑色油状液体,69%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ=6.99(t,j=10.0hz,2h,ph),6.87(d,j=8.7hz,2h,ph),6.71(d,j=8.7hz,2h,ph),6.59
–
6.52(m,3h,ph),3.49(t,j=10.0hz,2h,ch
2-n),1.56
–
1.46(m,2h,ch2),1.31
–
1.26(m,2h,ch2),0.84(t,j=7.4hz,3h,ch3)。
[0079]
化合物4(4d):实施例1方法,淡黄色固体,53%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4):δ=7.09(t,j=7.9hz,2h,ph),6.97(d,j=8.4hz,2h,ph),6.81(d,
j=8.5hz,2h,ph),6.66(d,j=8.1hz,3h,ph),3.58(t,j=7.6hz,2h,ch
2-n),1.63(t,j=7.4hz,2h,ch2),1.32
–
1.27(m,22h,c
11h22
),0.92(t,j=6.7hz,3h,ch3)。
[0080]
化合物5(4e):实施例1方法,白色固体,75%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ=7.31(d,j=7.17hz,2h,ph),7.25(t,j=7.65hz,2h,ph),7.17(t,j=7.14hz,1h,ph),7.07
–
7.01(m,4h,ph),6.77(d,j=8.83hz,2h,ph),6.71
–
6.68(m,2h,ph),6.65(t,j=7.29hz,1h,ph),4.84(s,2h,ch2)。
[0081]
化合物6(4f):实施例1方法,淡黄色色固体,81%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ=7.41(d,j=1.9hz,1h,h-furan),7.13
–
7.09(m,2h,ph),7.02(d,j=8.8hz,2h,ph),6.79(t,j=8.6hz,4h,ph),6.70(tt,j=5hz,1h,h-furan),6.31(dd,j=3.2,1.9hz,1h,h-furan),6.17(dd,j=3.2,1.9hz,1h,ph),4.79(s,2h,ch2)。
[0082]
化合物7(4g):实施例1方法,淡黄色色固体,66%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4):δ=8.07(d,j=9.0hz,1h,naphthalene),7.89(d,j=7.5hz,1h,naphthalene),7.74(d,j=8.2hz,1h,naphthalene),7.56
–
7.47(m,3h,ph),7.35(t,j=7.7hz,1h,naphthalene),7.12
–
7.03(m,4h,ar),6.78
–
6.66(m,5h,ar),5.30(s,2h,ch2)。
[0083]
化合物8(4h):实施例1方法,黑色固体,47%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ=7.15
–
7.05(m,4h,ph),7.04
–
6.95(m,2h,ph),6.91
–
6.78(m,2h,ph),6.75
–
6.65(m,3h,ph),6.65
–
6.56(m,3h,ph),3.81(t,j=6.9hz,2h,ch
2-nh),3.35
–
3.32(m,2h,ch
2-n)。
[0084]
化合物9(4i):实施例1方法,黑色液体,70%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ=7.23
–
7.16(m,2h,ph),7.11
–
7.05(m,2h,ph),6.63(d,j=8.9hz,2h,ph),6.45(d,j=8.9hz,2h,ph),3.13(s,3h,ch
3-n),2.08(s,3h,ch3)。
[0085]
化合物10(4j):实施例1方法,黑色液体,81%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ=7.02(t,j=7.8hz,1h,ph),6.96(d,j=8.7hz,2h,ph),6.80(d,j=8.8hz,2h,ph),6.58
–
6.50(m,3h,ph),3.18(s,3h,ch
3-n),2.22(s,3h,ch3)。
[0086]
化合物11(4j):实施例1方法,棕色固体,91%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ=6.97(d,j=8.2hz,2h,ph),6.92(d,j=8.8hz,2h,ph),6.77(d,j=8.8hz,2h,ph),6.68(d,j=8.5hz,2h,ph),3.16(s,3h,ch
3-n),2.23(s,3h,ch3)。
[0087]
化合物12(4j):实施例1方法,灰色固体,77%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ=6.92(d,j=8.68hz,2h,ph),6.87(t,j=8.80hz,2h,ph),6.77(d,j=8.78hz,2h,ph),6.73
–
6.69(m,2h,ph),3.16(s,3h,ch
3-n)。
[0088]
化合物13(4m):实施例1方法,灰色固体,62%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ=7.05(q,j=5.0hz,1h,ph),6.99(d,j=8.7hz,2h,ph),6.81(d,j=8.7hz,2h,ph),6.41(dd,j=8.3,2.4hz,1h,ph),6.36
–
6.28(m,2h,ph),3.18(d,j=0.9hz,3h,ch
3-n)。
[0089]
化合物14(4n):实施例1方法,黑色油状,63%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ=7.45(d,j=8.0hz,1h,ph),7.31(t,j=7.6hz,1h,ph),7.23(d,j=8.0hz,1h,ph),7.17(t,j=7.6hz,1h,ph),6.68(d,j=8.9hz,2h,ph),6.56(d,j=8.9hz,2h,ph),3.19(s,3h,ch
3-n)。
[0090]
化合物15(4o):实施例1方法,淡黄色固体,68%收率。核磁波谱数据:1h nmr
(500mhz,methanol-d4)δ=7.08
–
7.03(m,2h,ph),6.99
–
6.94(m,2h,ph),6.81(d,j=8.8hz,2h,ph),6.64
–
6.60(m,2h,ph),3.16(d,j=4.7hz,3h,ch
3-n)。
[0091]
化合物16(4p):实施例1方法,黑色固体,63%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ=7.66(d,j=8.0hz,1h,ph),7.36(t,j=7.6hz,1h,ph),7.22(d,j=8.0hz,1h,ph),7.11(t,j=7.6hz,1h,ph),6.68(d,j=8.9hz,2h,ph),6.53(d,j=8.8hz,2h,ph),3.18(s,3h,ch
3-n)。
[0092]
化合物17(4q):实施例1方法,黄色固体,41%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4,ph)δ=8.01(d,j=9.4hz,2h,ph),7.06(d,j=8.7hz,2h,ph),6.88(d,j=8.8hz,2h,ph),6.63(d,j=9.5hz,2h,ph),3.34(s,3h,ch
3-n)。
[0093]
化合物18(4r):实施例1方法,淡黄色固体,37%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ=7.58
–
7.49(m,2h,ph),7.16(d,j=8.4hz,1h,ph),7.01(t,j=7.5hz,1h,ph),6.92(d,j=8.9hz,2h,ph),6.78(d,j=8.8hz,2h,ph),3.34(s,3h,ch
3-n)。
[0094]
化合物19(4s):实施例1方法,黑色液体,71%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4):δ=7.15(t,j=8.07,1h,ph),7.04(dd,j=10.40,2h,ph),6.92(t,j=7.81,1h,ph),6.62(d,j=8.95hz,2h,ph),6.54(d,j=8.95hz,2h,ph),3.73(s,3h,ch
3-o),3.11(s,3h,ch
3-n)。
[0095]
化合物20(4t):实施例1方法,淡黄色固体,93%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4):δ=6.82-6.76(m,6h,ph),6.73-6.69(m,2h,ph),3.71(s,3h,ch
3-o),3.12(s,3h,ch
3-n)。
[0096]
化合物21(4u):实施例1方法,淡黄色液体,74%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ=7.44(d,j=7.60hz,1h,ph),7.37(t,j=7.61hz,1h,ph),7.29(t,j=7.51hz,1h,ph),7.24(d,j=7.90hz,1h,ph),6.68(d,j=9.02hz,2h,ph),6.58
–
6.53(m,4h,ph),6.41(t,j=2.30hz,1h,ph),3.66(s,6h,ch
3-o),2.87(s,3h,ch
3-n)。
[0097]
化合物22(4v):实施例1方法,黑色液体,63%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4):δ=7.35(d,j=7.63,1h,ph),7.23-7.16(m,2h,ph),7.01(d,j=7.7hz,1h,ph),6.62-6.59(m,2h,ph),6.41-6.37(m,2h,ph),3.20-3.14(m,1h,ch),3.11(s,3h,ch
3-n),1.13(d,j=6.9hz,6h,ch3)。
[0098]
化合物23(4w):实施例1方法,黑色固体,71%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ=6.88
–
6.78(m,6h,ph),6.72(d,j=8.8hz,2h,ph),5.99
–
5.90(m,1h,ch=c),5.23(dd,j=17.2,1.9hz,1h,ch2=c),5.12(dd,j=10.3,1.8hz,1h,ch2=c),4.23(dt,j=5.2,1.7hz,2h,ch
2-c=c),3.75(s,3h,ch
3-o)。
[0099]
化合物24(4x):实施例1方法,棕色液体,53%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ=7.24(d,j=8.7hz,1h,ar),7.02(s,1h,ar),6.92
–
6.61(m,10h,ar),3.84(d,j=7.6hz,2h,ch
2-n),3.76(d,j=10.9hz,6h,ch
3-o),3.01(d,j=7.9hz,2h,ch
2-indole)。
[0100]
化合物25(4y):实施例1方法,淡黄色固体,72%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ=7.47
–
7.42(m,2h,ph),7.35
–
7.27(m,3h,ph),7.10(td,j=8.1,4.3hz,1h,ph),7.03
–
6.96(m,2h,ph),6.86
–
6.79(m,4h,ph),6.67(dt,j=6.7,3.2hz,1h,ph),3.19(d,j=5.9hz,3h,ch
3-n)。
[0101]
化合物26(4z):实施例1方法,紫色固体,53%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ=7.92
–
7.83(m,2h,ar),7.69(d,j=8.2hz,1h,ar),7.48
–
7.41(m,2h,ar),7.35(t,j=7.3hz,1h,ar),7.26(d,j=7.3hz,1h,ar),6.62(d,j=8.9hz,2h,ar),6.54(d,j=9.0hz,2h,ar),3.31(s,3h,ch3)。
[0102]
化合物27(4aa):实施例1方法,白色固体,88%收率。核磁波谱数据:1h nmr(400mhz,chloroform-d)δ=7.37
–
7.15(m,4h,ph),7.13
–
6.89(m,8h,ph),6.77(d,j=8.1hz,2h,ph),4.73(s,1h,oh)。
[0103]
化合物28(4ab):实施例3方法,棕色固体,65%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ7.01(d,j=8.8hz,2h,ph),6.80(d,j=8.7hz,2h,ph),6.75-6.70(m,1h,ph),6.61-6.54(m,2h,ph),6.53-6.46(m,1h,ph),4.19(t,2h,ch
2-o),3.51(t,2h,ch
2-n)。
[0104]
化合物29(4ac):实施例4方法,白色固体,83%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ=7.09(d,j=8.7hz,2h,ph),6.99(d,j=8.6hz,2h,ph),6.88(dd,j=7.4,1.6hz,2h,ph),6.72(m,4h,ph),6.14(dd,j=8.2,1.3hz,2h,ph)。
[0105]
化合物30(4ad):实施例3方法,白色固体,72%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ6.97(d,j=8.8hz,2h,ph),6.88(d,j=9.0hz,1h,ph),6.78(d,j=8.7hz,2h,ph),6.74(t,j=8.5hz,1h,ph),6.50(t,j=7.3hz,1h,ph),6.32(d,j=9.3hz,1h,ph),3.47-3.39(m,2h,ch
2-n),2.74(t,j=6.4hz,2h,ch
2-ph),1.98-1.84(m,2h,ch2)。
[0106]
化合物31(4ae):实施例6方法,红色固体,79%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ=6.97
–
6.91(m,3h,ph),6.87
–
6.80(m,1h,ph),6.77
–
6.70(m,2h,ph),6.65(d,j=7.9hz,1h,ph),6.53(t,j=7.3hz,1h,ph),3.56(t,j=8.4hz,2h,ch
2-n),2.83(t,j=8.4hz,2h,ch
2-ph)。
[0107]
化合物32(7a):实施例2方法,红色固体,90%收率。核磁波谱数据:1h nmr(400mhz,methanol-d4)δ6.86-6.81(m,2h,ph),6.76-6.66(m,2h,ph),3.79(t,j=4.0hz,4h,c2),2.97(t,j=4.0hz,4h,c3)。
[0108]
化合物33(7b):实施例2方法,灰色固体,88%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ6.85(d,j=9.0hz,2h,ph),6.67(d,j=8.8hz,2h,ph),2.91(t,4h,c1),1.74-1.63(m,4h,c2),1.56-1.43(m,2h,c3)。
[0109]
化合物34(7c):实施例2方法,白色色固体,82%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ7.35(d,j=8.3hz,2h,ph),7.03(d,j=8.4hz,2h,ph),3.96-3.47(m,4h,c2),3.16(d,j=6.8hz,4h,c3)。
[0110]
化合物35(10a):实施例8方法,黑色液体,71%收率。核磁波谱数据:1h nmr(500mhz,chloroform-d)δ6.94(d,j=8.8hz,1h,ph),6.85-6.77(m,3h,ph),6.72-6.66(m,3h,ph),5.81(s,1h,oh),3.76(s,3h,ch
3-o),3.14(s,3h,ch
3-n),3.00-2.91(m,4h,c2),1.69(m,j=5.8hz,c3),1.51(t,j=6.0hz,2h,c4)。
[0111]
化合物36(10b):实施例8方法,黑色液体,62%收率。核磁波谱数据:1h nmr(500mhz,chloroform-d)δ7.23(t,j=7.8hz,2h,ph),6.99(d,j=8.9hz,1h,ph),6.86-6.81(m,2h,ph),6.72(d,j=8.2hz,2h,ph),6.68(d,j=2.8hz,1h,ph),5.71(s,1h,oh),3.82(t,j=4.7hz,4h,c2),3.20(s,3h,ch
3-n),3.01(t,j=4.7hz,4h,c3)。
[0112]
化合物38(10c):实施例8方法,黑色液体,78%收率。核磁波谱数据:1h nmr(500mhz,chloroform-d)δ6.96(d,j=8.7hz,1h,ph),6.84-6.75(m,3h,ph),6.73-6.61(m,3h,ph),5.87(s,1h,oh),3.81(d,j=4.6hz,4h,c2),3.75(s,3h,ch
3-o),3.14(s,3h,ch
3-n),2.99(t,j=4.8hz,4h,c3)。
[0113]
化合物37(10d):实施例8方法,红色液体,47%收率。核磁波谱数据:1h nmr(500mhz,chloroform-d)δ6.98(d,j=8.8hz,1h,ph),6.91(t,j=8.7hz,2h,ph),6.81(dd,j=8.9,2.9hz,1h,ph),6.70-6.60(m,3h,ph),5.78(s,1h,oh),3.98-3.53(m,4h,c2),3.16(s,3h,ch
3-n),3.06-2.92(m,4h,c3)。
[0114]
化合物39(10e):实施例8方法,黄色固体,61%收率。核磁波谱数据:1h nmr(500mhz,methanol-d4)δ7.05(d,j=9.1hz,2h,ph),6.88(d,j=8.9hz,1h,ph),6.80(dd,j=8.9,2.9hz,1h,ph),6.69(d,j=3.0hz,1h,ph),6.56(d,j=9.1hz,2h,ph),3.81-3.67(m,4h,c2),3.15(s,3h,ch
3-n),2.99-2.83(m,4h,c3)。
[0115]
癸氨基苯酚:白色固体,产率98%。核磁波谱数据:1h nmr(400mhz,chloroform-d)δ6.66(d,j=8.2hz,2h,ph),6.54(d,j=8.2hz,2h,ph),4.37(br,2h,oh&nh),3.04(t,j=7.2hz,2h,c1),1.59(t,j=7.3hz,2h,c2),1.43
–
1.19(m,14h,c3-c9),0.89(t,j=6.7hz,3h,c10)。
[0116]
下面对上述实施例制备的部分化合物进行体外抗菌试验、mtt细胞活性测定、酪氨酸酶抑制试验、酪氨酸酶抑制的动力学分析以及抗氧化试验。
[0117]
一、体外抗菌试验
[0118]
我们对本发明上述实施例提供的部分化合物进行体外抗菌试验。
[0119]
方法:根据国家临床实验室标准委员会的方法测定mic。
[0120]
选择3株革兰氏阳性菌和3株革兰氏阴性菌作为试验菌。蜡状芽孢杆菌和大肠杆菌购自中国普通微生物菌种保藏管理中心。mrsa(金黄色葡萄球菌atcc 43300)和枯草芽孢杆菌由西北农林科技大学化学与药学院提供。白菜软腐和魔芋软腐由西北农林科技大学植物保护学院提供。mic被定义为最小抑制浓度,每种化合物对细菌生长产生明显抑制(在37℃下培养12-14h)。将每种细菌悬浮液的浓度调整为1
×
106cfu/ml。称重前,将所有化合物彻底干燥。最初,将化合物溶解在二甲基亚砜(dmso)中以制备储备溶液。然后在液体luriae bertan培养基中制备受试化合物和对照药物。所需浓度分别为128、64、32、16、8、4、2、1、0.5和0.25μg/ml(二甲基亚砜《0.5%)。选择环丙沙星和庆大霉素作为阳性对照(k.p.rakesh et al.2020.
[1]
)。
[0121]
本实验采用二倍稀释法对40种参试化合物的最小抑制浓度(mic值)进行测定。在无菌条件下向5ml离心管中加入3ml灭菌后的lb培养液,再用灭菌的接种针挑取少量供试细菌加入到培养液中,封好口后于37.5℃恒温培养箱中培养12h。取100μl灭菌的培养液于600nm下测定吸光度,以不加供试细菌的lb培养液为空白对照,根据0.1od600相当于浓度为1
×
108cfu/ml,后将菌液稀释100倍至1
×
106cfu/ml。使用96孔(u型底,12
×
8)微孔稀释板进行mic测定。先将化合物配置成51200μg/ml的dmso溶液备用,吸取5μl溶液,加入无菌水1995μl,配制成初始浓度为128μg/ml的母液。在第2-8孔中各加入100μl的无菌水,在第1孔加入200μl母液,将第1孔中溶液混合均匀后用移液枪移取100μl至第2孔,以此类推对2-8孔进行二倍稀释,最后从第8孔中吸出混合好的100μl溶液弃掉,如此便可获得128~1μg/ml浓度梯
度。向1-8和10孔中各加入菌液100μl,如此1-8孔便可获得64~0.5μg/ml浓度梯度。10、11孔中加入100μl的无菌水,11孔加入100μl的lb培养液,12孔加入200μl无菌水作为空白对照。
[0122]
将微孔中溶液震荡均匀,置于37℃恒温培养箱中培养;以庆大霉素及环丙沙星作为阳性对照,每个处理重复三次(阳性对照初始浓度与药液浓度同为64μg/ml)。对mics初测为0.5μg/ml的化合物采用同样操作进一步测试其在0.5~0.015μg/ml浓度区间内的最低抑制浓度值。观察培养12-16h后的结果,有菌丝生长的孔中会有沉淀,呈现出混浊状,无菌丝生长的孔的药液浓度即为该样品的mic值。
[0123]
供试细菌:蜡状芽孢杆菌(bacillus cereus,1.1846)、枯草芽孢杆菌(bacillus subtilis,1.88)、耐甲氧西林金黄色葡萄球菌(staphylococcus aureus,1.89)、大肠杆菌(escherichia coli,1.1574),白菜软腐病菌(erwinia carotovora.a)及魔芋软腐病菌(erwinia carotovora.b)。阳性对照药物:环丙沙星,庆大霉素。
[0124]
表1抗菌活性数据
[0125][0126][0127]
a mrsa=methcillin-resistant staphylococcus aureus(耐甲氧西林金黄色葡萄球菌),b.cereus=bacillus cereus(蜡状芽孢杆菌),b.subtilis=bacillus subtilis
(枯草芽孢杆菌),e.coli=escherichia coli(大肠杆菌)。
[0128]
b erwinia carotovora subsp.carotovora isolated from cabbage.(胡萝卜欧文氏菌亚种。从卷心菜中分离出的菌种)。
[0129]
c erwinia carotovora subsp.carotovora isolated from konjak.(胡萝卜欧文氏菌亚种。从魔芋中分离出的菌种)。
[0130]
d dap=p-decylaminophenol(对癸氨基苯酚)。
[0131]
e cip=ciprofloxacin(环丙沙星),gent=gentamicin.(庆大霉素)。
[0132]
如表1所示,本发明实施例1~8制备的大多数化合物对革兰氏阳性菌(尤其是耐甲氧西林金黄色葡萄球菌)具有抗菌活性,对革兰氏阴性菌的抗菌活性较低。其中,对mrsa具有抗菌活性的化合物的结构通式如下式a1和a2所示:
[0133][0134]
式a1和a2中,r1选自-me、-et、-f、-cl、-br、-ome、-ipr、-cn、-no2中的任意一种;r2选自-ph或h;x选自c、o、s、n中的任意一种;n=0或1。
[0135]
在上述化合物中,化合物4k,4m,4n,4p和4ab是抗mrsa最有效的化合物,其最小mic值为0.5μg/ml,接近阳性对照药物(mic
cip
=0.25μg/ml;mic
gen
=0.25μg/ml)。化合物4k,4o和4ab尽管mic值高于阳性对照,但仍显示出一定的蜡样芽孢杆菌抑制活性,mic值为16μg/ml;就抗枯草芽孢杆菌而言,化合物4m表现出较好的活性(mic=8μg/ml),显示出潜在的衍生前景;所有化合物对革兰氏阴性菌活性较弱,最低mic值为64μg/ml。
[0136]
二、mtt细胞活性测定
[0137]
我们对本发明实施例1~8制备的部分化合物进行mtt细胞活性测定。
[0138]
其中,式a1和a2所示的化合物的mtt细胞活性数据基本相同,因此,下面仅以化合物(4k,4m,4n,4p,4ab)来说明其mtt细胞活性。
[0139]
方法:受试化合物(4k,4m,4n,4p,4ab)的细胞活力根据duan
[2]
等人描述的方法进行测定(j.duan et al.2016.
[2]
)。针对raw 264.7细胞系检测抗mrsa活性的化合物(浓度为0.5、1、2、4、8、16和32μg/ml),每个化合物进行3个重复的体外细胞活性测试,以确定的潜在毒性作用。对照细胞单独加入二甲基亚砜,其浓度等于药物处理细胞样品中的浓度。具有不同浓度和细胞的化合物在37℃的5%co2中在96孔板中培养12h。移除培养基,添加0.5mg/ml mtt培养4h。去除培养基。然后向每个孔中添加dmso(100μl)。震动15min,以溶解紫色甲臜晶体。最后,选取波长为570nm该板进行测试。空白组作为对照组。数据通过graphpad prism 6.0进行分析。用于细胞活力的公式为(处理细胞的吸光度/未处理细胞的吸光度)
×
100%。
[0140]
供试细胞:raw 264.7细胞
[0141]
使用mtt法进一步评估生物活性,化合物4k,4m,4n,4p,4ab对raw264.7细胞的毒性,结果如图1所示。
[0142]
由图1结果可知,mtt法检测raw 264.7细胞系中4k,4m,4n,4p和4ab的细胞毒性(误差条代表吸光度值的标准偏差值。通过student’s t-test.检验,与对照组相比,*p《
0.05、**p《0.01或***p《0.001。)
[0143]
化合物与细胞培养24h后,在0.5μg/ml浓度下,4k,4m,4n,4p,4ab的细胞存活率超过95%。而化合物4k,4m,4n在有效抗菌剂量(0.5μg/ml)下对raw 264.7细胞表现出极好的存活率(105%、103%和103%)。化合物4k,4m,4n,4p和4ab在8μg/ml浓度下对raw 264.7细胞无毒。实验结果表明所有受试化合物在有效浓度下对细胞的毒性较低。
[0144]
三、酪氨酸酶抑制试验
[0145]
我们对本发明上述实施例提供的部分化合物进行酪氨酸酶抑制试验。
[0146]
对酪氨酸酶活性具有抑制效果的化合物通式如下式a3和a4所示:
[0147][0148]
式a3和a4中,r3选自me、f、cl、br中的任意一种。
[0149]
式a3和a4所示的化合物对酪氨酸酶活性的抑制效果基本相同,因此,下面仅以化合物(4k,4m,4n,4p,4ab)来说明其对酪氨酸酶活性的抑制效果。
[0150]
方法:蘑菇酪氨酸酶(ec 1.14.18.1)(sigma chemical co.)使用l-酪氨酸作为底物进行酶抑制试验(v.n.takahashi et al.2014
[3]
)。在dmso(40mm)中制备供试化合物4k,4m,4n,4p和4ab以及曲酸的储备溶液,然后用磷酸盐缓冲液(ph=6.8)稀释至所需浓度。首先,将10μl蘑菇酪氨酸酶(0.5mg/ml)与160μl磷酸盐缓冲液(50mm,ph=6.8)在96孔微孔板中混合,然后添加10μl试验化合物。将混合物在28℃下预培养20min后,向每个孔中添加20μl的l-酪氨酸溶液(0.5mm),并在475nm处监测10min。所有受试化合物最终浓度为16μm。每孔重复三次,将不含试验化合物的dmso用作对照。试验溶液中dmso的最终浓度小于2.0%。根据以下方程式计算抑制率:
[0151]
抑制率(%)=100
×
(测试化合物)/对照
[0152]
每种化合物的抑制效果表示为酶活性抑制50%所需的浓度(ic50)。
[0153]
供试酶:蘑菇酪氨酸酶(ec 1.14.18.1)
[0154]
为了验证n-芳基对胺基苯酚的抗mrsa活性与酪氨酸酶抑制活性之间的关系,我们选择有良好抑菌活性的化合物4k,4m,4n,4p和4ab来完成实验。我们使用蘑菇酪氨酸酶作为被抑制酶,曲酸(ka)作为阳性对照,所有化合物在最终浓度为16μm的条件下处理(m.d.aytemir et al.2019
[4]
)。当使用l-酪氨酸作为反应底物时,所有化合物均表现出显著良好的抗酪氨酸酶活性。
[0155]
图2是化合物对酪氨酸酶的影响结果。其中,(a)是各种化合物在16μm时对酪氨酸酶活性的影响。结果代表各组的平均值
±
sd。通过student’s t-test检验,与对照组相比,p《0.05、**p《0.01或***p《0.001。(b)是化合物对蘑菇酪氨酸酶活性的浓度依赖效应(平均值)。
[0156]
由图2结果可知,与对照组相比,4k,4m,4n,4p,4ab和ka分别抑制了47%、39%、44%、49%、53%和26%的酪氨酸酶活性。我们还研究了不同浓度的4k,4m,4n,4p和4ab对蘑菇酪氨酸酶活性的影响,并计算了5种化合物的ic50值。这些化合物都显示出比ka更高的活
性。化合物4k,4m,4n,4p和4ab的ic50值分别为3.95μm、2.65μm、2.56μm、3.35μm和2.53μm。这些结果表明,此类具有高酪氨酸酶抑制活性的化合物也具有良好的抗mrsa活性,这其中,4ab是蘑菇酪氨酸酶活性最有效的抑制剂。
[0157]
四、酪氨酸酶抑制的动力学分析
[0158]
我们对本发明上述实施例提供的部分化合物进行酪氨酸酶抑制的动力学分析。
[0159]
方法:化合物4ab的抑制动力学中,我们选择浓度为:0、1、2、4、8、16和32μm。底物l-酪氨酸浓度为0、0.062、0.125、0.25、0.5、1、2和4mm,在所有动力学研究中伴随酪氨酸酶的浓度为0.5mg/ml。预培养和测量时间与蘑菇酪氨酸酶抑制试验方案中的方法相同。抑制类型通过速度(1/v)与底物浓度(1/[s])的倒数的lineweavere-burk图进行分析。解离常数ki由斜率与抑制剂浓度的二次曲线图确定。
[0160]
采用lineweaver-burk双倒数法分析了最有效的化合物4ab的抑制机制。在存在不同抑制剂浓度的情况下,通过绘制1/v与1/[s]的曲线对酶进行动力学研究,得出了一系列在第二象限横穿x轴的直线,如图3所示。当化合物4ab的浓度升高时,vmax值降低,而km值不变,这表明4ab对蘑菇酪氨酸酶的抑制类型是非竞争性的。
[0161]
图3是在化合物4ab存在下酪氨酸酶氧化l-酪氨酸的动力学图。其中,(a)是在0、1、2、4、8、16和32μm浓度下,3-1j抑制酪氨酸酶的lineweaver-burk图。底物l-酪氨酸的浓度分别为0.0625、0.125、0.25、0.5、1、2和4μm。(b)是斜率与抑制剂(4ab)浓度的曲线图。
[0162]
通过斜率与化合物4ab浓度的二次曲线图测定离解常数ki(图3)。从这条直线上看,4ab对酶的抑制常数ki为1.76μm,见表2。
[0163]
表2酶抑制动力学参数
[0164][0165]
注:nda表示未测。
[0166]
五、抗氧化试验
[0167]
我们对本发明实施例1~8制备的部分化合物进行抗氧化试验。实施例1~8制备的化合物均具有相应的抗氧化效果,因此,下面仅以化合物(4k,4m,4n,4p,4ab)来说明其抗氧化效果。
[0168]
方法:根据sarker
[5]
等人描述的方法测量受试化合物的dpph清除活性,并进行了一些修改(sarker et al.2005
[5]
)。用移液管将100ml不同浓度(3.125、6.25、12.5、25、50和100μg/ml)的受试化合物(4k,4m,4n,4p和4ab)移到96孔平底板中。接下来,向每个孔中添加100mm的dpph甲醇溶液100μl,并在室温下对平板进行避光培养30min。在λ517nm处测量溶液的吸光度(v.padmavathi et al.2011
[6]
)。抗坏血酸和二甲基亚砜分别作为阳性对照和空白。根据以下方程式计算dpph清除活性的百分比:
[0169]
%dpph清除率=[(a
空白-a
样品
)/a
空白
]
×
100
[0170]
其中空白为对照反应的吸光度(包含除试验化合物外的所有试剂),样品为试验样品的吸光度。在自由基清除活性%和药物浓度之间绘制剂量-反应曲线。对显示50%自由基
抑制活性(ic50)的药物浓度进行线性回归分析。抗坏血酸作为阳性对照,所有试验在三个重复中进行,结果取平均值。
[0171]
阳性对照:抗坏血酸。选取具有良好抗菌活性的化合物在不同浓度(3.125、6.25、12.5、25、50和100μg/ml)下进行抗氧化活性评估,并与标准抗氧化剂(抗坏血酸)进行比较研究。我们选择dpph(1,1-二苯基-2-吡啶酰肼)自由基清除活性评估方法作为抗氧化活性研究的标准分析方法。结果显示在图4中。图4中,抗坏血酸、4k,4m,4n,4p和4ab对dpph的清除作用百分比(%)。每个值代表平均值
±
标准差(n=3)。
[0172]
由图4结果表明,4-(2,3-二氢-4h-苯并[b][1,4]恶嗪-4-基)苯酚(化合物4ab)(ic50=7.82μg/ml)显示出比其他对应物更高的抗氧化活性潜力,是抗坏血酸的1.24倍(ic50=9.70μg/ml)。化合物4k(ic50=10.45μg/ml),4m(ic50=10.22μg/ml),4n(ic50=14.04μg/ml)和4p(ic50=12.57μg/ml显示出与阳性对照相当的抗氧化活性。
[0173]
六、结果与分析
[0174]
通过对筛选出的5个化合物均表现出较低的细胞毒性、良好的抗氧化活性以及酪氨酸酶抑制活性,其中,化合物4n和4ab酪氨酸酶抑制活性表现优秀,ic50值分别为2.65μm和2.53μm。此5个化合物生物活性见表3。
[0175]
表3化合物的生物活性
[0176][0177]
注:nd表示未测。
[0178]
综上,针对n-单烷基取代的对胺基苯酚通常不稳定易被氧化及衍生结构单一的问题,本发明通过引入n-芳基对胺基酚使结构稳定,增强化合物可衍生性,对此类结构的抗菌、抗氧化和酪氨酸抑制活性进行评价,找到了若干同时具有酪氨酸酶抑制活性和抗菌活性的化合物。本发明具有化合物合成简单,针对mrsa(耐甲氧西林金黄色葡萄球菌)抑菌效
果好,酶抑制率高等特点,有着广阔的应用前景。
[0179]
引用文献:
[0180]
[1]h.qin,j.liu,w.fang,l.ravindar,k.p.rakesh,indole-based deriv atives as potential antibacterial activity against methicillin-resistance staphyloco ccus aureus(mrsa),eur.j.med.chem.194(2020)112245-112261.
[0181]
[2]h.mu,q.liu,n.hong,y.sun,j.duan,gold nanoparticles make chitosan-streptomycin conjugates effective towards gram-negative bacterial biofil m.rsc advances,11(2016)8714-8721.
[0182]
[3]n.takahashi,m.imai,k.yu,inhibitory effects ofp-alkylaminopheno l on melanogenesis,bioorg.med.chem.22(17)(2014)4677-4683.
[0183]
[4]g.karakaya,a.t
ü
re,a.ercan,s.m.d.aytemir,synthesis,computational molecular docking analysis and effectiveness on tyrosinase inhibiti on ofkojic acid derivatives.bioorg.chem.88(2019)102950.
[0184]
[5]l.nahar,w.r.russell,m.middleton,m.shoeb,s.d.sarker,antioxi dant phenylacetic acid derivatives from the seeds ofilex aquifolium,acta pharm aceutica 55(2005)187-193,uri.
[0185]
[6]a.padmaja,c.rajasekhar,a.muralikrishna,v.padmavathi,synthesi s and antioxidant activity ofoxazolyl/thiazolylsulfonylmethyl pyrazoles and isox azoles,eur.j.med.chem.46(10)(2011)5034-5038.
[0186]
以上仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1